Training a custom scAdam model#
[1]:
# Python packages
import warnings
warnings.simplefilter('ignore')
import scanpy as sc
import scparadise
import seaborn as sns
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
sc.set_figure_params(dpi = 120)
Recommendations about training dataset#
We recommend shifted logarithm data normalization method: sc.pp.normalize_total(adata, target_sum=None) sc.pp.log1p(adata) But you can use any other method of data normalzation (Use the same normalization method for test dataset) Training dataset sould contain all genes that you want to use for model training in adata_train.X We recommend to remove all non-marker genes from adata_train.X (removeing of such useless genes increase performance and model quality metrics)
Data preparation#
Here we download and preprocess dataset from cellxgene:
Single cell RNA sequencing of oropharyngeal squamous cell carcinoma https://cellxgene.cziscience.com/collections/3c34e6f1-6827-47dd-8e19-9edcd461893f
[2]:
!wget https://datasets.cellxgene.cziscience.com/915069db-1df2-49a1-9a9c-2fbd0aa13c81.h5ad
--2025-01-16 12:42:52-- https://datasets.cellxgene.cziscience.com/915069db-1df2-49a1-9a9c-2fbd0aa13c81.h5ad
Resolving datasets.cellxgene.cziscience.com (datasets.cellxgene.cziscience.com)... 52.85.49.125, 52.85.49.28, 52.85.49.24, ...
Connecting to datasets.cellxgene.cziscience.com (datasets.cellxgene.cziscience.com)|52.85.49.125|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 981941987 (936M) [binary/octet-stream]
Saving to: ‘915069db-1df2-49a1-9a9c-2fbd0aa13c81.h5ad’
915069db-1df2-49a1- 100%[===================>] 936.45M 4.91MB/s in 2m 16s
2025-01-16 12:45:08 (6.89 MB/s) - ‘915069db-1df2-49a1-9a9c-2fbd0aa13c81.h5ad’ saved [981941987/981941987]
[3]:
# Load prepared for training anndata object
adata = sc.read_h5ad('915069db-1df2-49a1-9a9c-2fbd0aa13c81.h5ad')
[4]:
# Get raw counts from adata.raw
adata = adata.raw.to_adata()
[5]:
# Convert var_names from ENSG codes to gene names
adata.var.set_index('feature_name', inplace=True)
adata.var_names_make_unique()
[6]:
# Create celltype_l1 and celltype_l2 annotation levels
adata.obs['celltype_l2'] = adata.obs['cell_type'].copy()
# Check current cluster name
cluster_list = adata.obs['celltype_l2'].unique()
# Make cluster anottation dictionary
annotation = {
"Epithelial": ['epithelial cell'],
"B": ['B cell', 'plasma cell'],
"T": ['CD4-positive, alpha-beta T cell', 'CD8-positive, alpha-beta T cell', 'mature alpha-beta T cell', 'regulatory T cell'],
"NK": ['natural killer cell'],
"Myeloid": ['myeloid cell', 'plasmacytoid dendritic cell', 'mast cell'],
"Endothelial": ['endothelial cell', 'endothelial cell of lymphatic vessel'],
"Stromal": ['fibroblast', 'mural cell']
}
# Change dictionary format
annotation_rev = {}
for i in cluster_list:
for k in annotation:
if i in annotation[k]:
annotation_rev[i] = k
adata.obs["celltype_l1"] = [annotation_rev[i] for i in adata.obs['celltype_l2']]
[7]:
# Check annotations
sc.pl.umap(
adata,
color = [
'celltype_l1',
'celltype_l2'
],
frameon = False,
ncols = 1
)
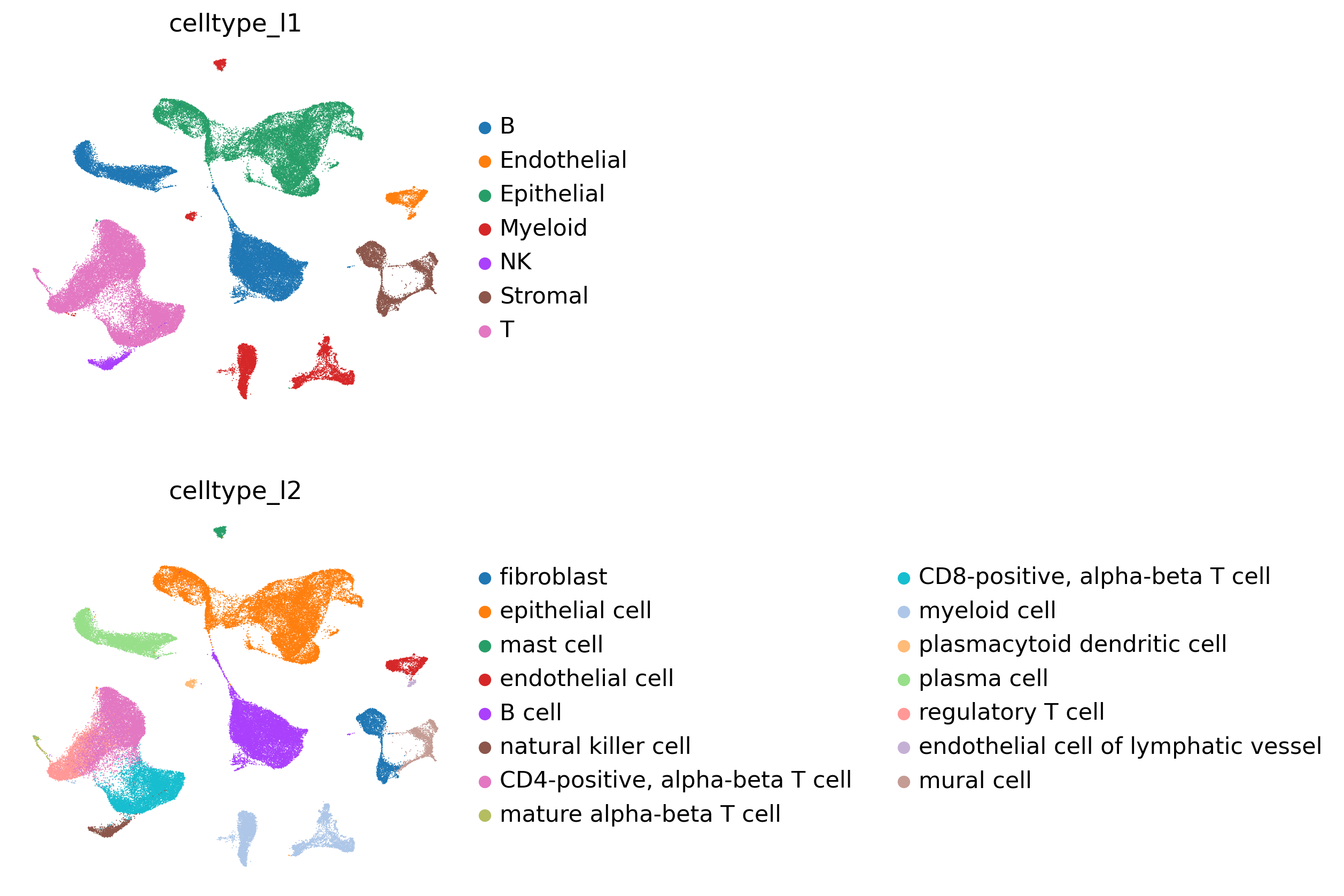
[8]:
# Create adata_train1, adata_train2 and adata_test datasets
adata_test = adata[adata.obs['donor_id'].isin(['HN481', 'HN482'])].copy()
adata_train = adata[adata.obs['donor_id'].isin(['HN483' 'HN485', 'HN488', 'HN489', 'HN490', 'HN487', 'HN492', 'HN494'])].copy()
del adata
[9]:
# Normalize data
for i in [adata_train, adata_test]:
i.layers['counts'] = i.X.copy()
sc.pp.normalize_total(i, target_sum=None)
sc.pp.log1p(i)
[10]:
# Find highly variable features for scAdam model training
sc.pp.highly_variable_genes(
adata_train,
layer='counts',
flavor='seurat_v3',
n_top_genes=1200,
subset=True
)
Balanced training#
We recommend balance dataset based on most detailed annotation level (last in celltype_keys list - celltype_l2 in this case). Balancing the training dataset increases the sensitivity (balanced accuracy), f1-score and geometric mean of the model but leads to slightly decrease in precision
[11]:
# Balance dataset based on most detailed annotation level
adata_balanced = scparadise.scnoah.balance(
adata_train,
celltype_keys = ['celltype_l1', 'celltype_l2']
)
[12]:
# Train scadam model using adata_balanced dataset
scparadise.scadam.train(
adata_balanced,
path = '', # path to save model
model_name = 'model_scadam_balanced', # folder name with model
celltype_keys = [
'celltype_l1', # First (less detailed) annotation level
'celltype_l2', # Second (most detailed) annotation level
],
eval_metric = ['accuracy', 'balanced_accuracy']
)
Device: cuda
Number of features: 1200
Label hierarchy: celltype_l1 → celltype_l2
Annotation levels weights using strategy 'linear_offset':
celltype_l1: 7 cell types, 0.4 relative weight
celltype_l2: 15 cell types, 0.6 relative weight
Dataset split:
Train dataset contains: 36903 cells, it is 80.0 % of input dataset
Validation dataset contains: 9226 cells, it is 20.0 % of input dataset
Unsupervised pretraining: 100%|█████████████████████████████████████████████████████████| 50/50 [18:46<00:00, 22.54s/it]
Training scAdam model: 16%|█████████ | 32/200 [17:40<1:32:47, 33.14s/it]
Early stopping triggered! Best score: 0.9805
Training completed!
Fitting unknown cells detector
UnknownCellDetector fitted successfully!
Model saved to model_scadam_balanced
Check model quality#
[13]:
# Predict cell types using trained model
adata_test = scparadise.scadam.predict(
adata_test,
path_model = 'model_scadam_balanced'
)
scAdam model with unknown detector loaded from model_scadam_balanced
Gene alignment:
Model features: 1200
Matched features: 1200 (100.0%)
Predicting: 100%|███████████████████████████████████████████████████████████████████████| 72/72 [00:01<00:00, 67.99it/s]
Added cell type column: pred_celltype_l1
Added probabilities column: pred_celltype_l1_probability
Added cell type column: pred_celltype_l2
Added probabilities column: pred_celltype_l2_probability
[14]:
# Add prediction status to adata_test
for i in ['l1', 'l2']:
scparadise.scnoah.pred_status(
adata_test,
celltype = 'celltype_' + i,
pred_celltype = 'pred_celltype_' + i,
key_added = 'pred_status_' + i
)
[15]:
# Order cell type colors
for i in ['l1', 'l2']:
celltype = np.unique(adata_test.obs['celltype_' + i]).tolist()
adata_test.obs['celltype_' + i] = pd.Categorical(
values=adata_test.obs['celltype_' + i], categories=celltype, ordered=True
)
adata_test.obs['pred_celltype_' + i] = pd.Categorical(
values=adata_test.obs['pred_celltype_' + i], categories=celltype, ordered=True
)
[16]:
# Visualise predicted cell types (celltype_l1) and prediction probabilities
sc.pl.umap(
adata_test,
color=[
'celltype_l1', # observed cell type annotations
'pred_celltype_l1', # predicted cell type annotations
'pred_celltype_l1_probability' # prediction probabilities
],
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 7,
legend_fontoutline = 1,
ncols = 3,
wspace = 0.2,
hspace = 0.1
)
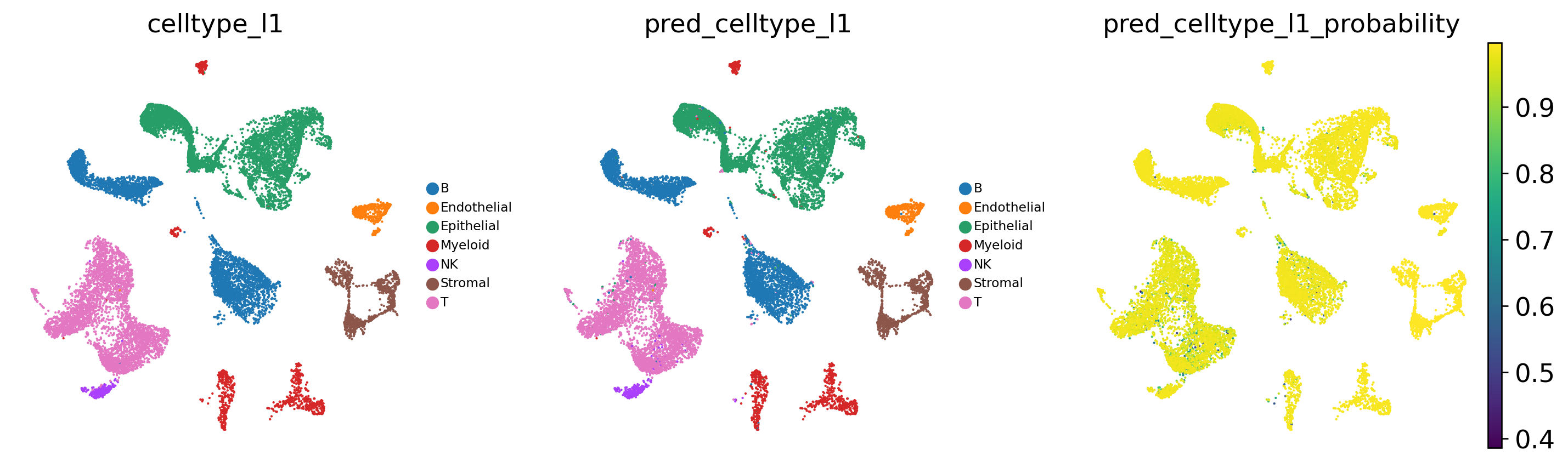
[17]:
# Visualise predicted cell types (celltype_l2) and prediction probabilities
sc.pl.umap(
adata_test,
color=[
'celltype_l2', # observed cell type annotations
'pred_celltype_l2', # predicted cell type annotations
'pred_celltype_l2_probability' # prediction probabilities
],
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 5,
legend_fontoutline = 1,
ncols = 2,
wspace = 0.6,
hspace = 0.1
)
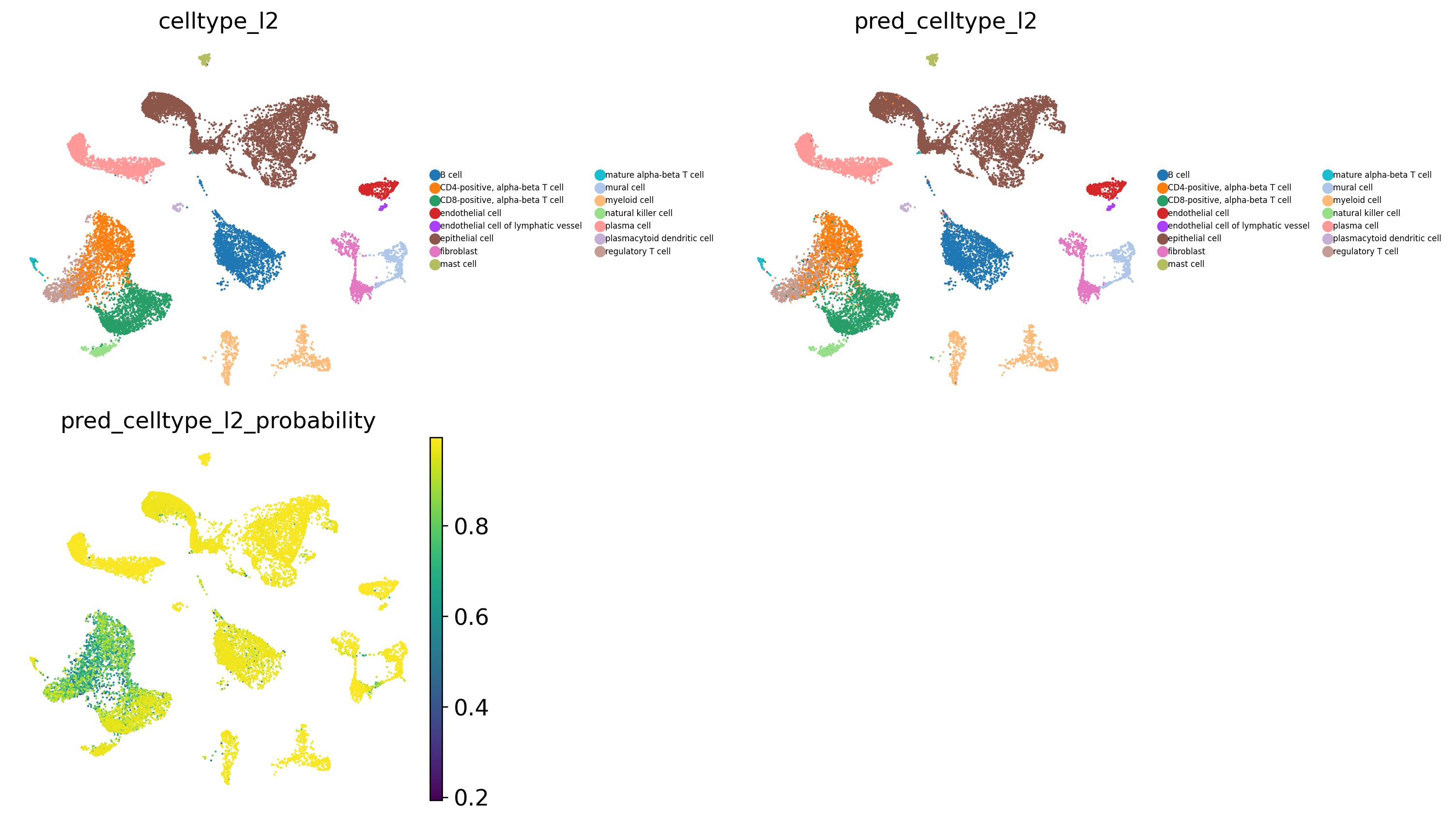
[18]:
# Visualise prediction status
sc.pl.embedding(
adata_test,
color=[
'pred_status_l1',
'pred_status_l2'
],
basis = 'X_umap',
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 7,
legend_fontoutline = 1,
ncols = 3,
wspace = 0.2,
hspace = 0.1
)
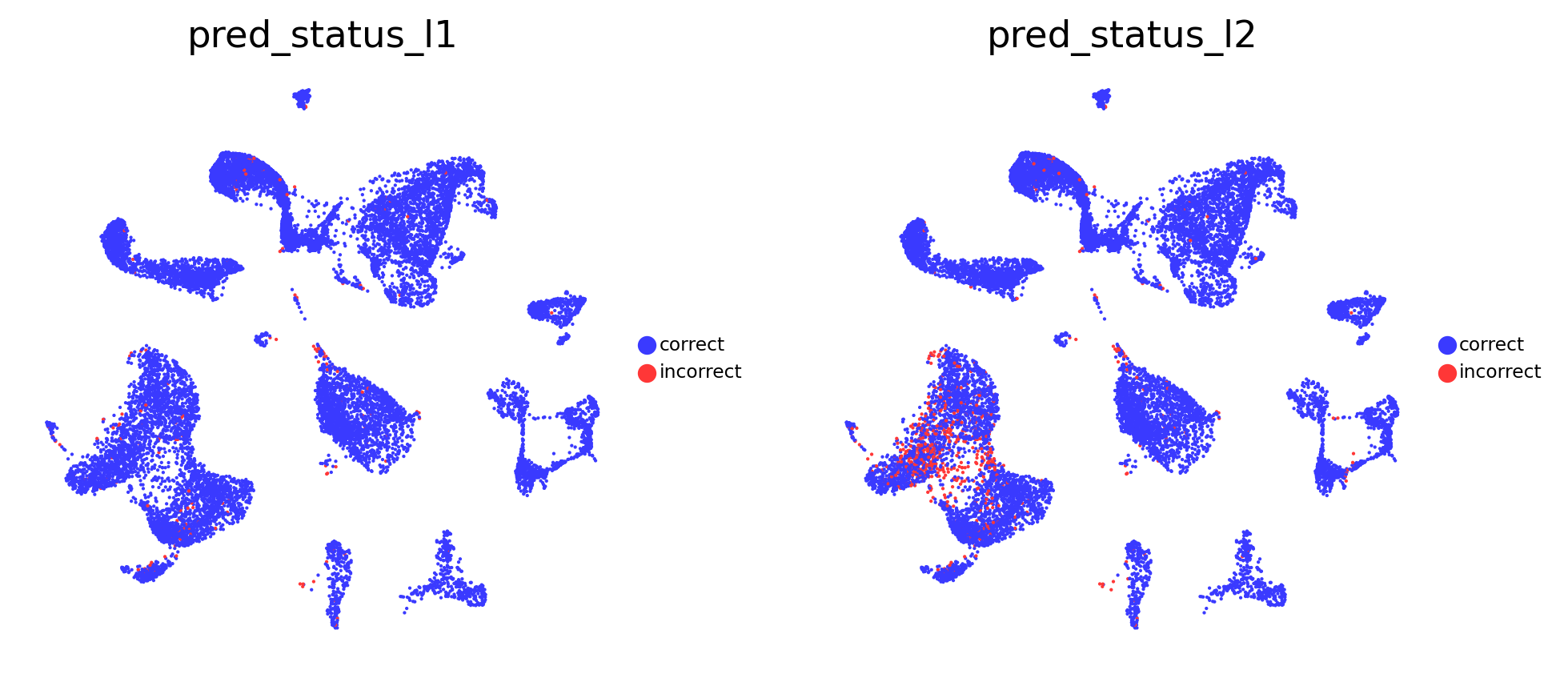
The probability and prediction status analysis indicates issues with the model in annotating T cell subtypes at the celltype_l2 annotation level. However, the model does not have problems with annotating other cell types, as confirmed by the quality analysis presented below.
Comparizon of observed and predicted cell type annotations#
To compare the actual and predicted cell types, we use report_classif_full and conf_matrix from the scNoah module. More information about the metrics in scparadise.scnoah.report_classif_full is available in the scParadise documentation. More information about confusion matrix (scparadise.scnoah.conf_matrix) is available
here.
[19]:
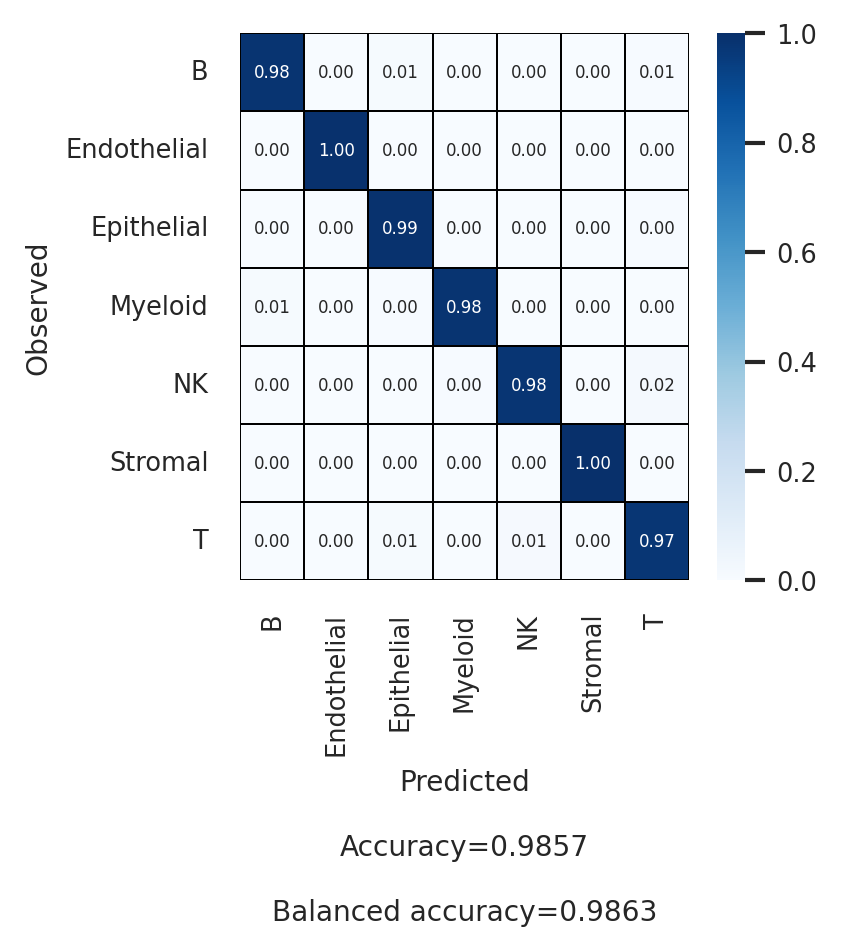
# First annotation level (celltype_l1)
df_l1 = scparadise.scnoah.report_classif_full(
adata_test,
celltype = 'celltype_l1',
pred_celltype = 'pred_celltype_l1',
ndigits = 3
)
df_l1
[19]:
| precision | recall/sensitivity | specificity | f1-score | geometric mean | index balanced accuracy | number of cells | |
|---|---|---|---|---|---|---|---|
| B | 0.992 | 0.983 | 0.998 | 0.987 | 0.99 | 0.979 | 3969 |
| Endothelial | 0.998 | 0.996 | 1.0 | 0.997 | 0.998 | 0.996 | 509 |
| Epithelial | 0.990 | 0.991 | 0.993 | 0.991 | 0.992 | 0.984 | 7505 |
| Myeloid | 0.966 | 0.982 | 0.998 | 0.974 | 0.99 | 0.978 | 924 |
| NK | 0.804 | 0.978 | 0.997 | 0.883 | 0.987 | 0.973 | 223 |
| Stromal | 0.995 | 1.0 | 1.0 | 0.997 | 1.0 | 1.0 | 1113 |
| T | 0.985 | 0.975 | 0.996 | 0.98 | 0.985 | 0.968 | 4057 |
| macro avg | 0.961 | 0.986 | 0.997 | 0.973 | 0.992 | 0.983 | |
| weighted avg | 0.986 | 0.986 | 0.996 | 0.986 | 0.991 | 0.98 | |
| Accuracy | 0.986 | ||||||
| Balanced accuracy | 0.986 |
[20]:
sns.set(font_scale = 0.7)
plt.figure(figsize = (3, 3))
scparadise.scnoah.conf_matrix(
adata_test,
celltype = 'celltype_l1',
pred_celltype = 'pred_celltype_l1',
annot_kws = {"size":5},
linewidths = 0.1, linecolor = 'black',
fmt = ".2f",
ndigits_metrics = 4,
vmin = 0, vmax = 1
)
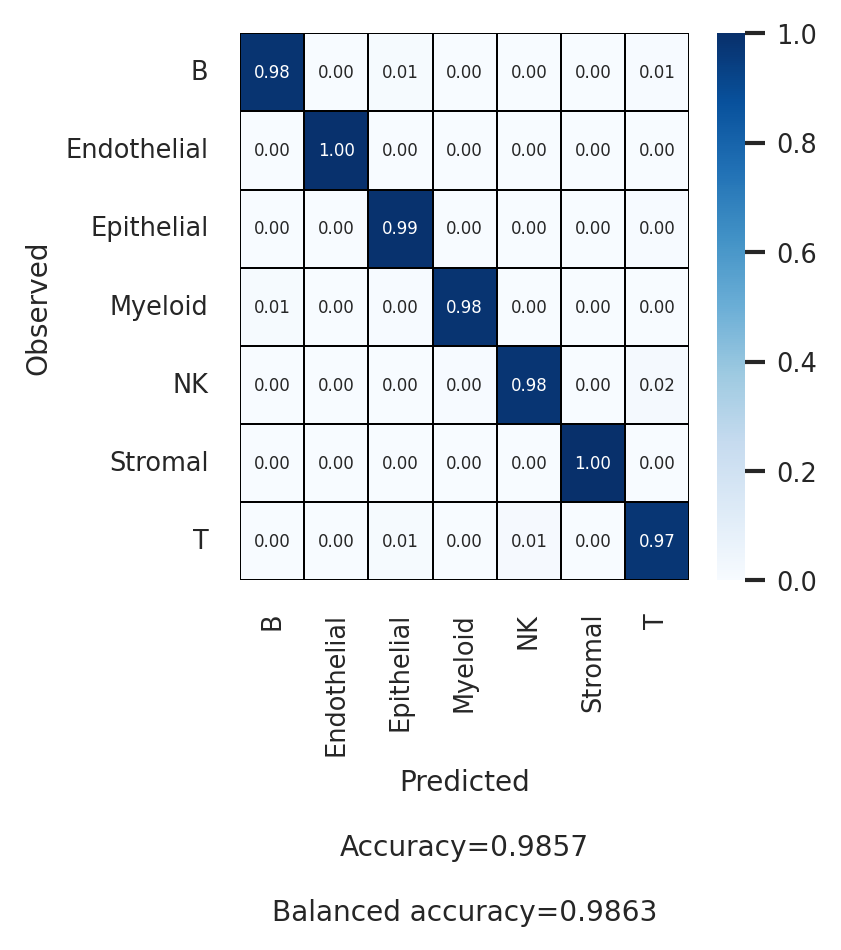
[21]:
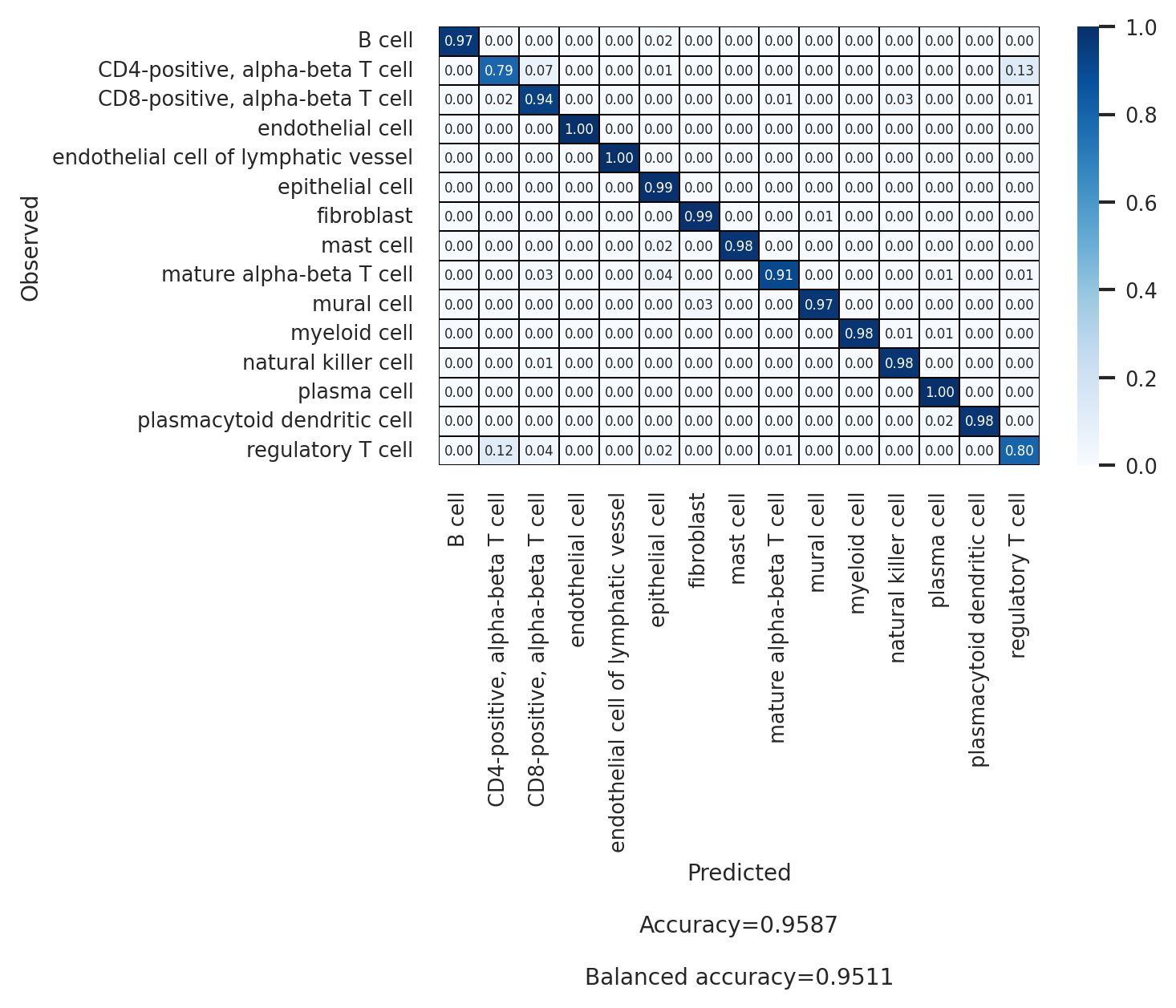
# Second annotation level (celltype_l2)
df_l2 = scparadise.scnoah.report_classif_full(
adata_test,
celltype = 'celltype_l2',
pred_celltype = 'pred_celltype_l2',
ndigits = 3
)
df_l2
[21]:
| precision | recall/sensitivity | specificity | f1-score | geometric mean | index balanced accuracy | number of cells | |
|---|---|---|---|---|---|---|---|
| B cell | 0.994 | 0.966 | 0.999 | 0.98 | 0.983 | 0.963 | 2050 |
| CD4-positive, alpha-beta T cell | 0.898 | 0.79 | 0.992 | 0.84 | 0.885 | 0.767 | 1554 |
| CD8-positive, alpha-beta T cell | 0.920 | 0.94 | 0.991 | 0.93 | 0.966 | 0.927 | 1742 |
| endothelial cell | 0.998 | 0.998 | 1.0 | 0.998 | 0.999 | 0.998 | 457 |
| endothelial cell of lymphatic vessel | 1.000 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 52 |
| epithelial cell | 0.989 | 0.992 | 0.992 | 0.99 | 0.992 | 0.984 | 7505 |
| fibroblast | 0.978 | 0.993 | 0.999 | 0.985 | 0.996 | 0.991 | 676 |
| mast cell | 0.980 | 0.98 | 1.0 | 0.98 | 0.99 | 0.978 | 148 |
| mature alpha-beta T cell | 0.773 | 0.907 | 0.999 | 0.834 | 0.952 | 0.897 | 75 |
| mural cell | 0.988 | 0.97 | 1.0 | 0.979 | 0.985 | 0.967 | 437 |
| myeloid cell | 0.981 | 0.978 | 0.999 | 0.979 | 0.989 | 0.975 | 726 |
| natural killer cell | 0.784 | 0.978 | 0.997 | 0.87 | 0.987 | 0.972 | 223 |
| plasma cell | 0.985 | 0.996 | 0.998 | 0.99 | 0.997 | 0.994 | 1919 |
| plasmacytoid dendritic cell | 0.961 | 0.98 | 1.0 | 0.97 | 0.99 | 0.978 | 50 |
| regulatory T cell | 0.705 | 0.799 | 0.987 | 0.749 | 0.888 | 0.774 | 686 |
| macro avg | 0.929 | 0.951 | 0.997 | 0.938 | 0.973 | 0.944 | |
| weighted avg | 0.960 | 0.959 | 0.994 | 0.959 | 0.976 | 0.95 | |
| Accuracy | 0.959 | ||||||
| Balanced accuracy | 0.951 |
[22]:
sns.set(font_scale = 0.7)
plt.figure(figsize = (5, 3))
scparadise.scnoah.conf_matrix(
adata_test,
celltype = 'celltype_l2',
pred_celltype = 'pred_celltype_l2',
annot_kws = {"size":5},
linewidths = 0.1, linecolor = 'black',
fmt = ".2f",
ndigits_metrics = 4,
vmin = 0, vmax = 1
)
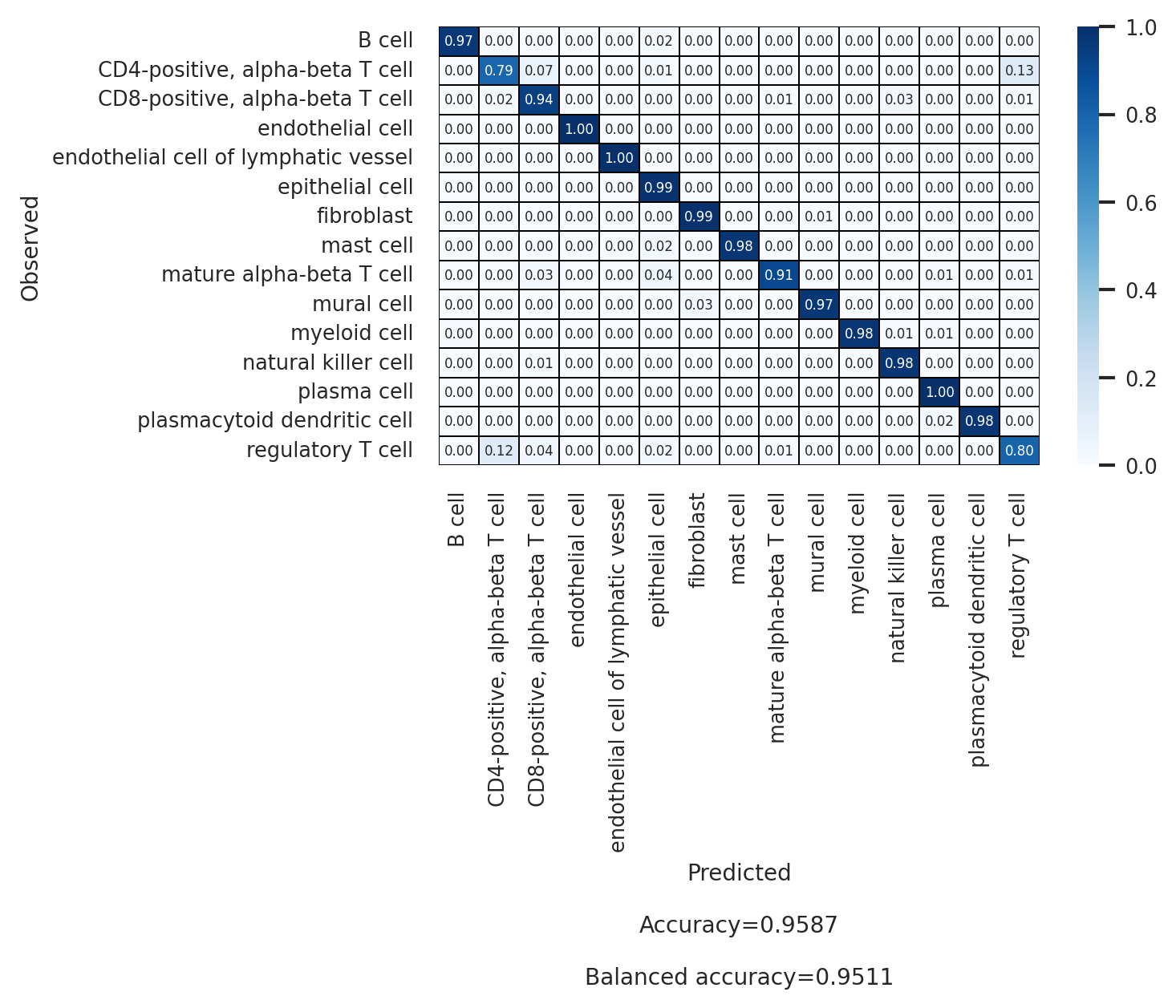
[23]:
# Save anndata with predicted annotations
adata_test.write_h5ad('adata_test_balanced_model.h5ad')
Imbalanced training#
Use ‘balanced_accuracy’ as evaluation metric in case of imbalanced learning
[24]:
# Train scadam model using adata_train dataset
scparadise.scadam.train(
adata_balanced,
path = '', # path to save model
model_name = 'model_scadam_imbalanced', # folder name with model
celltype_keys = [
'celltype_l1', # First (less detailed) annotation level
'celltype_l2', # Second (most detailed) annotation level
],
eval_metric = ['accuracy', 'balanced_accuracy']
)
Device: cuda
Number of features: 1200
Label hierarchy: celltype_l1 → celltype_l2
Annotation levels weights using strategy 'linear_offset':
celltype_l1: 7 cell types, 0.4 relative weight
celltype_l2: 15 cell types, 0.6 relative weight
Dataset split:
Train dataset contains: 36903 cells, it is 80.0 % of input dataset
Validation dataset contains: 9226 cells, it is 20.0 % of input dataset
Unsupervised pretraining: 100%|█████████████████████████████████████████████████████████| 50/50 [17:51<00:00, 21.44s/it]
Training scAdam model: 16%|█████████▍ | 32/200 [11:08<58:28, 20.89s/it]
Early stopping triggered! Best score: 0.9805
Training completed!
Fitting unknown cells detector
UnknownCellDetector fitted successfully!
Model saved to model_scadam_imbalanced
Check model quality#
[25]:
# Predict cell types using trained model
adata_test = scparadise.scadam.predict(
adata_test,
path_model = 'model_scadam_imbalanced'
)
scAdam model with unknown detector loaded from model_scadam_imbalanced
Gene alignment:
Model features: 1200
Matched features: 1200 (100.0%)
Predicting: 100%|███████████████████████████████████████████████████████████████████████| 72/72 [00:00<00:00, 74.50it/s]
Added cell type column: pred_celltype_l1
Added probabilities column: pred_celltype_l1_probability
Added cell type column: pred_celltype_l2
Added probabilities column: pred_celltype_l2_probability
[26]:
# Add prediction status to adata_test
for i in ['l1', 'l2']:
scparadise.scnoah.pred_status(
adata_test,
celltype = 'celltype_' + i,
pred_celltype = 'pred_celltype_' + i,
key_added = 'pred_status_' + i
)
[27]:
# Order cell type colors
for i in ['l1', 'l2']:
celltype = np.unique(adata_test.obs['celltype_' + i]).tolist()
adata_test.obs['celltype_' + i] = pd.Categorical(
values=adata_test.obs['celltype_' + i], categories=celltype, ordered=True
)
adata_test.obs['pred_celltype_' + i] = pd.Categorical(
values=adata_test.obs['pred_celltype_' + i], categories=celltype, ordered=True
)
[28]:
# Visualise predicted cell types (celltype_l1) and prediction probabilities
sc.pl.umap(
adata_test,
color=[
'celltype_l1', # observed cell type annotations
'pred_celltype_l1', # predicted cell type annotations
'pred_celltype_l1_probability' # prediction probabilities
],
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 7,
legend_fontoutline = 1,
ncols = 3,
wspace = 0.2,
hspace = 0.1
)
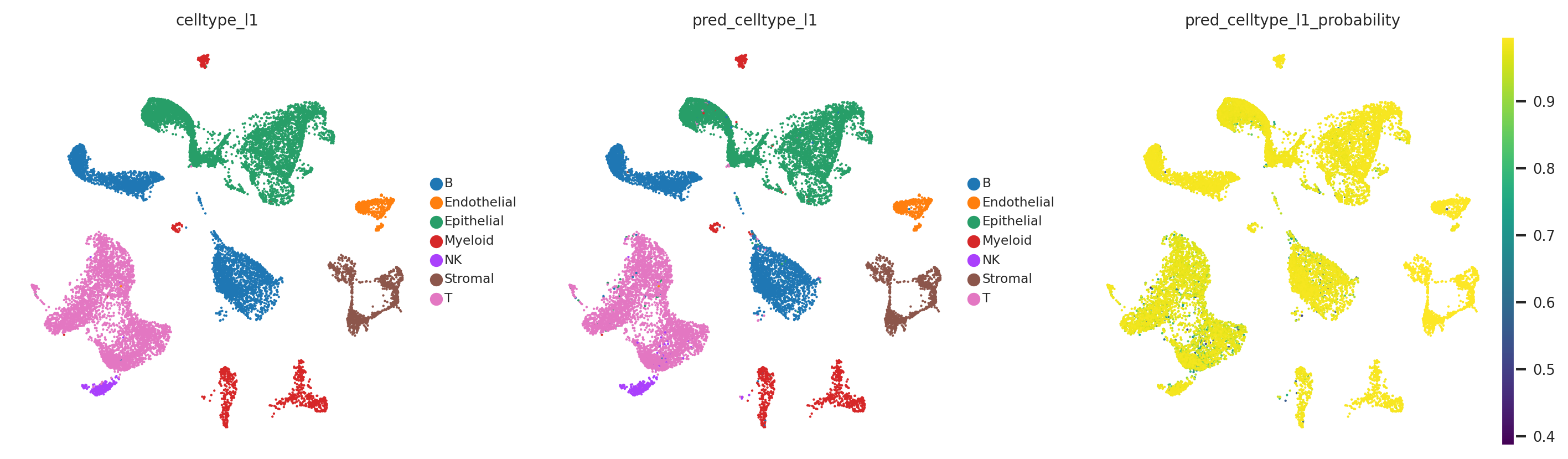
[29]:
# Visualise predicted cell types (celltype_l2) and prediction probabilities
sc.pl.umap(
adata_test,
color=[
'celltype_l2', # observed cell type annotations
'pred_celltype_l2', # predicted cell type annotations
'pred_celltype_l2_probability' # prediction probabilities
],
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 5,
legend_fontoutline = 1,
ncols = 2,
wspace = 0.5,
hspace = 0.1
)
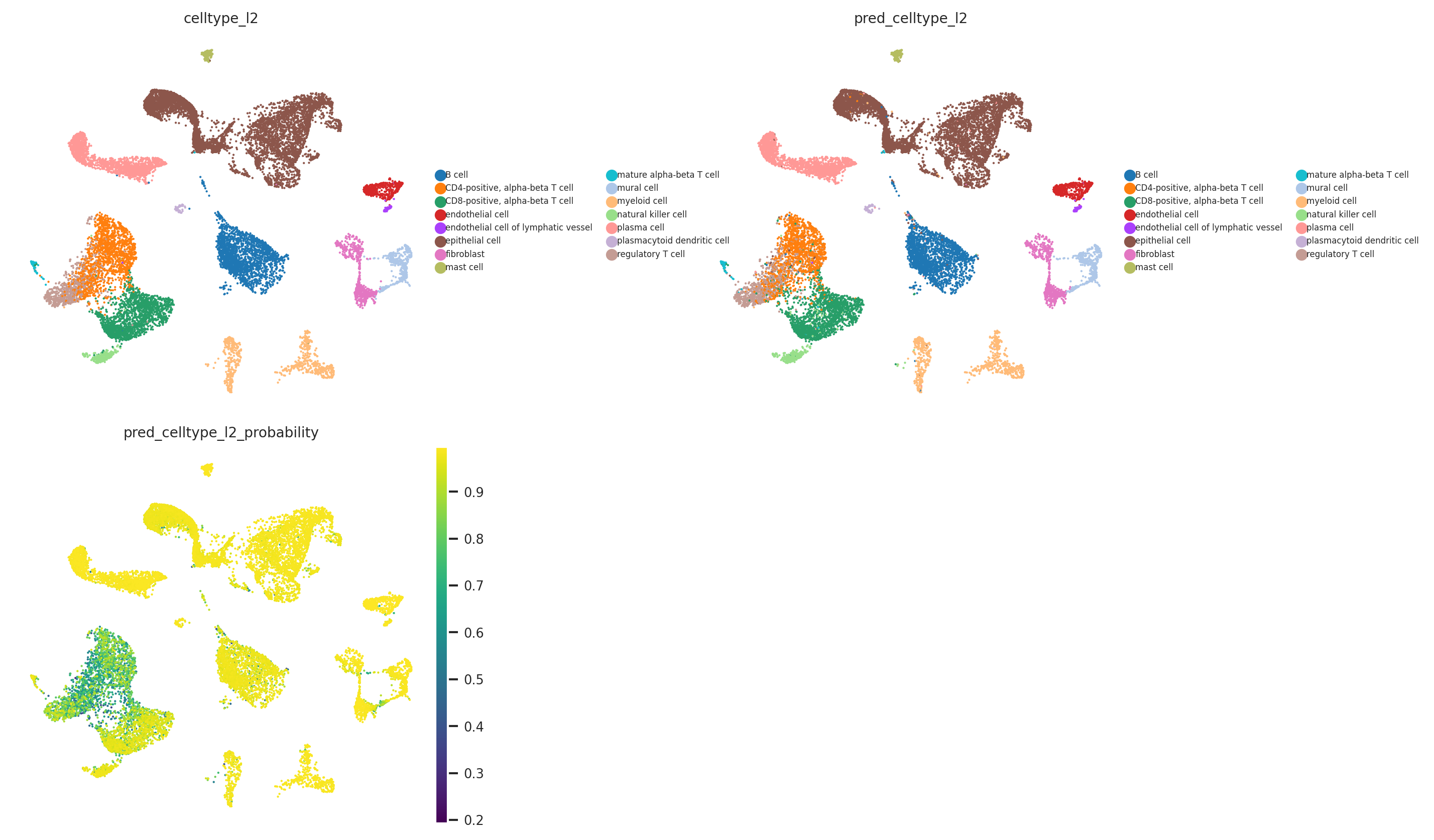
[30]:
# Visualise prediction status
sc.pl.embedding(
adata_test,
color=[
'pred_status_l1',
'pred_status_l2'
],
basis = 'X_umap',
frameon = False,
cmap = 'viridis',
legend_loc = 'right margin',
legend_fontsize = 7,
legend_fontoutline = 1,
ncols = 3,
wspace = 0.2,
hspace = 0.1
)
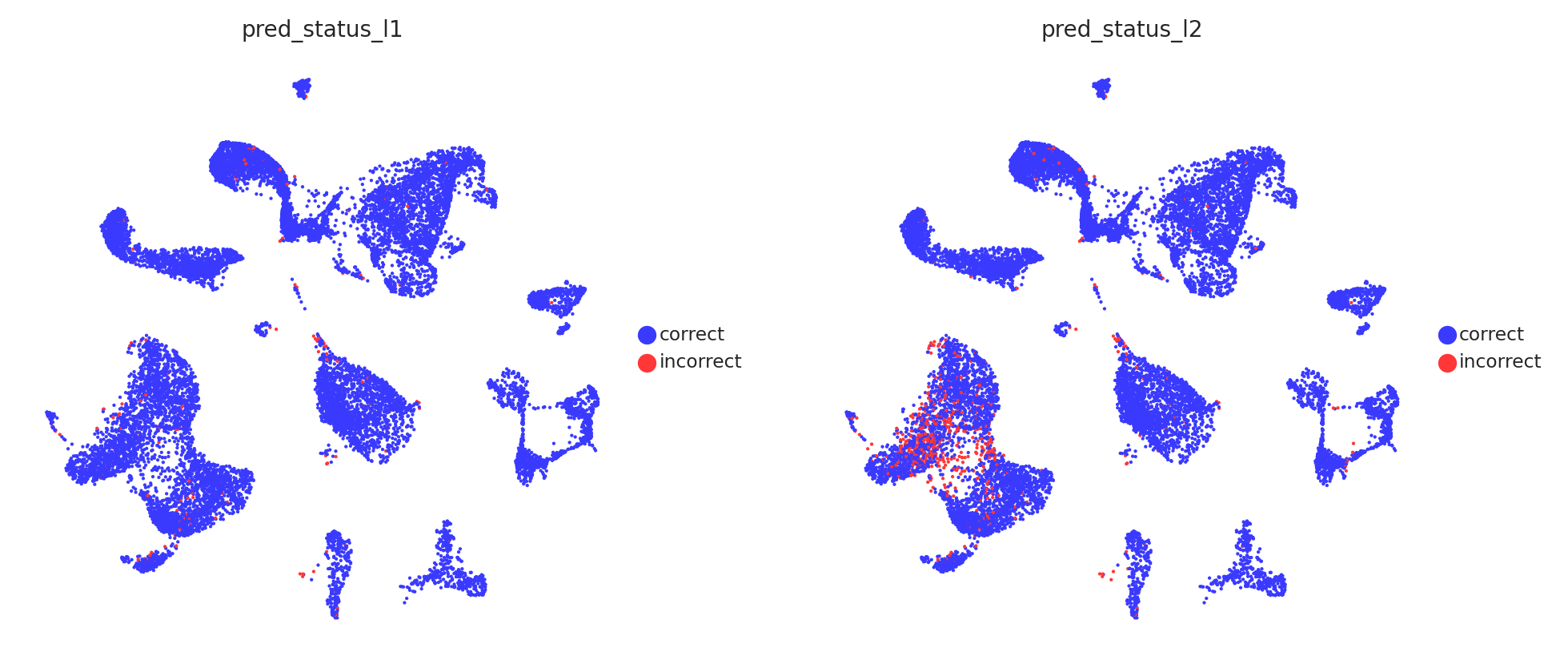
The probability and prediction status analysis indicates issues with the model in annotating T cell subtypes at the celltype_l2 annotation level. However, the model does not have problems with annotating other cell types, as confirmed by the quality analysis presented below.
Comparizon of observed and predicted cell type annotations#
To compare the actual and predicted cell types, we use report_classif_full and conf_matrix from the scNoah module. More information about the metrics in scparadise.scnoah.report_classif_full is available in the scParadise documentation. More information about confusion matrix (scparadise.scnoah.conf_matrix) is available
here.
[31]:
# First annotation level (celltype_l1)
df_l1 = scparadise.scnoah.report_classif_full(
adata_test,
celltype='celltype_l1',
pred_celltype='pred_celltype_l1',
ndigits = 3
)
df_l1
[31]:
| precision | recall/sensitivity | specificity | f1-score | geometric mean | index balanced accuracy | number of cells | |
|---|---|---|---|---|---|---|---|
| B | 0.992 | 0.983 | 0.998 | 0.987 | 0.99 | 0.979 | 3969 |
| Endothelial | 0.998 | 0.996 | 1.0 | 0.997 | 0.998 | 0.996 | 509 |
| Epithelial | 0.990 | 0.991 | 0.993 | 0.991 | 0.992 | 0.984 | 7505 |
| Myeloid | 0.966 | 0.982 | 0.998 | 0.974 | 0.99 | 0.978 | 924 |
| NK | 0.804 | 0.978 | 0.997 | 0.883 | 0.987 | 0.973 | 223 |
| Stromal | 0.995 | 1.0 | 1.0 | 0.997 | 1.0 | 1.0 | 1113 |
| T | 0.985 | 0.975 | 0.996 | 0.98 | 0.985 | 0.968 | 4057 |
| macro avg | 0.961 | 0.986 | 0.997 | 0.973 | 0.992 | 0.983 | |
| weighted avg | 0.986 | 0.986 | 0.996 | 0.986 | 0.991 | 0.98 | |
| Accuracy | 0.986 | ||||||
| Balanced accuracy | 0.986 |
[32]:
sns.set(font_scale = 0.7)
plt.figure(figsize = (3, 3))
scparadise.scnoah.conf_matrix(
adata_test,
celltype = 'celltype_l1',
pred_celltype = 'pred_celltype_l1',
annot_kws = {"size":5},
linewidths = 0.1, linecolor = 'black',
fmt = ".2f",
ndigits_metrics = 4,
vmin = 0, vmax = 1
)
[33]:
# Second annotation level (celltype_l2)
df_l2 = scparadise.scnoah.report_classif_full(
adata_test,
celltype='celltype_l2',
pred_celltype='pred_celltype_l2',
ndigits = 3
)
df_l2
[33]:
| precision | recall/sensitivity | specificity | f1-score | geometric mean | index balanced accuracy | number of cells | |
|---|---|---|---|---|---|---|---|
| B cell | 0.994 | 0.966 | 0.999 | 0.98 | 0.983 | 0.963 | 2050 |
| CD4-positive, alpha-beta T cell | 0.898 | 0.79 | 0.992 | 0.84 | 0.885 | 0.767 | 1554 |
| CD8-positive, alpha-beta T cell | 0.920 | 0.94 | 0.991 | 0.93 | 0.966 | 0.927 | 1742 |
| endothelial cell | 0.998 | 0.998 | 1.0 | 0.998 | 0.999 | 0.998 | 457 |
| endothelial cell of lymphatic vessel | 1.000 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 52 |
| epithelial cell | 0.989 | 0.992 | 0.992 | 0.99 | 0.992 | 0.984 | 7505 |
| fibroblast | 0.978 | 0.993 | 0.999 | 0.985 | 0.996 | 0.991 | 676 |
| mast cell | 0.980 | 0.98 | 1.0 | 0.98 | 0.99 | 0.978 | 148 |
| mature alpha-beta T cell | 0.773 | 0.907 | 0.999 | 0.834 | 0.952 | 0.897 | 75 |
| mural cell | 0.988 | 0.97 | 1.0 | 0.979 | 0.985 | 0.967 | 437 |
| myeloid cell | 0.981 | 0.978 | 0.999 | 0.979 | 0.989 | 0.975 | 726 |
| natural killer cell | 0.784 | 0.978 | 0.997 | 0.87 | 0.987 | 0.972 | 223 |
| plasma cell | 0.985 | 0.996 | 0.998 | 0.99 | 0.997 | 0.994 | 1919 |
| plasmacytoid dendritic cell | 0.961 | 0.98 | 1.0 | 0.97 | 0.99 | 0.978 | 50 |
| regulatory T cell | 0.705 | 0.799 | 0.987 | 0.749 | 0.888 | 0.774 | 686 |
| macro avg | 0.929 | 0.951 | 0.997 | 0.938 | 0.973 | 0.944 | |
| weighted avg | 0.960 | 0.959 | 0.994 | 0.959 | 0.976 | 0.95 | |
| Accuracy | 0.959 | ||||||
| Balanced accuracy | 0.951 |
[34]:
sns.set(font_scale = 0.7)
plt.figure(figsize = (5, 3))
scparadise.scnoah.conf_matrix(
adata_test,
celltype = 'celltype_l2',
pred_celltype = 'pred_celltype_l2',
annot_kws = {"size":5},
linewidths = 0.1, linecolor = 'black',
fmt = ".2f",
ndigits_metrics = 4,
vmin = 0, vmax = 1
)
[35]:
# Save anndata with predicted annotations
adata_test.write_h5ad('adata_test_imbalanced_model.h5ad')
[38]:
import session_info
session_info.show()
[38]:
Click to view session information
----- anndata 0.11.4 matplotlib 3.10.8 numpy 2.2.6 pandas 2.3.3 scanpy 1.11.5 scparadise 1.0.0 seaborn 0.13.2 session_info v1.0.1 -----
Click to view modules imported as dependencies
81d243bd2c585b0f4821__mypyc NA PIL 12.1.1 anyio NA arrow 1.4.0 asttokens NA attr 26.1.0 attrs 26.1.0 babel 2.18.0 certifi 2026.02.25 cffi 2.0.0 charset_normalizer 3.4.6 cloudpickle 3.1.2 colorlog NA comm 0.2.3 cuda 12.9.4 cycler 0.12.1 cython_runtime NA dateutil 2.9.0.post0 debugpy 1.8.20 decorator 5.2.1 defusedxml 0.7.1 exceptiongroup 1.3.1 executing 2.2.1 fastjsonschema NA fqdn NA fsspec 2026.2.0 h5py 3.16.0 idna 3.11 imblearn 0.14.1 ipykernel 7.2.0 isoduration NA jedi 0.19.2 jinja2 3.1.6 joblib 1.5.3 json5 0.13.0 jsonpointer 3.1.0 jsonschema 4.26.0 jsonschema_specifications NA jupyter_events 0.12.0 jupyter_server 2.17.0 jupyterlab_server 2.28.0 kiwisolver 1.5.0 lark 1.3.1 lazy_loader 0.5 legacy_api_wrap NA llvmlite 0.46.0 markupsafe 3.0.3 matplotlib_inline 0.2.1 mpl_toolkits NA mpmath 1.3.0 mudata 0.3.3 muon 0.1.7 natsort 8.4.0 nbformat 5.10.4 numba 0.64.0 optuna 4.8.0 overrides NA packaging 25.0 parso 0.8.6 patsy 1.0.2 pexpect 4.9.0 platformdirs 4.9.4 plottable 0.1.5 prometheus_client NA prompt_toolkit 3.0.52 psutil 7.2.2 ptyprocess 0.7.0 pure_eval 0.2.3 pycparser 3.00 pydev_ipython NA pydevconsole NA pydevd 3.2.3 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.19.2 pynndescent 0.6.0 pyparsing 3.3.2 pythonjsonlogger NA pytorch_tabnet NA pytz 2026.1.post1 referencing NA requests 2.32.5 rfc3339_validator 0.1.4 rfc3986_validator 0.1.1 rfc3987_syntax NA rpds NA scipy 1.15.3 send2trash NA shap 0.49.1 six 1.17.0 skimage 0.25.2 sklearn 1.7.2 sklearn_compat 0.1.5 skmisc 0.5.1 slicer NA stack_data 0.6.3 statsmodels 0.14.6 sympy 1.14.0 threadpoolctl 3.6.0 torch 2.10.0+cu128 torchgen NA tornado 6.5.5 tqdm 4.67.3 traitlets 5.14.3 triton 3.6.0 typing_extensions NA umap 0.5.11 uri_template NA urllib3 2.6.3 wcwidth 0.6.0 webcolors NA websocket 1.9.0 yaml 6.0.3 zmq 27.1.0 zoneinfo NA
----- IPython 8.38.0 jupyter_client 8.8.0 jupyter_core 5.9.1 jupyterlab 4.5.6 ----- Python 3.10.20 (main, Mar 11 2026, 17:46:40) [GCC 14.3.0] Linux-6.6.87.2-microsoft-standard-WSL2-x86_64-with-glibc2.39 ----- Session information updated at 2026-03-22 13:32