Cross-species modality prediction using scEve#
[1]:
import scanpy as sc
import scparadise
import os
from scipy import sparse
from mousipy import translate
import muon as mu
import numpy as np
import pandas as pd
import warnings
warnings.simplefilter('ignore')
sc.set_figure_params(dpi = 150, dpi_save = 600)
C:\Users\vadim\anaconda3\envs\scrna\lib\site-packages\tqdm\auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Recommendations about dataset#
Our models are trained using shifted logarithm normalized data. We recommend the shifted logarithm data normalization method for the proper use of our models. sc.pp.normalize_total(adata, target_sum=None) sc.pp.log1p(adata) adata.raw = adata
[2]:
# Load ADT data from 10x Genomics
url = "https://cf.10xgenomics.com/samples/cell-exp/7.2.0/4plex_mouse_LymphNode_Spleen_TotalSeqC_multiplex_LymphNode1_BC1_AB1/4plex_mouse_LymphNode_Spleen_TotalSeqC_multiplex_LymphNode1_BC1_AB1_count_sample_filtered_feature_bc_matrix.h5"
mdata = mu.read_10x_h5("dataset.h5", backup_url = url)
mdata.var_names_make_unique()
# RNA data
adata = mdata.mod['rna'].copy()
# Protein data
adata_adt = mdata.mod['prot'].copy()
del mdata
print('RNA data', adata, sep='\n')
print('Protein data', adata_adt, sep='\n')
RNA data
AnnData object with n_obs × n_vars = 7579 × 19059
var: 'gene_ids', 'feature_types', 'genome'
Protein data
AnnData object with n_obs × n_vars = 7579 × 122
var: 'gene_ids', 'feature_types', 'genome'
Gene expression data processing#
[3]:
# mitochondrial genes, "MT-" for human, "Mt-" for mouse
adata.var["mt"] = adata.var_names.str.startswith("mt-")
[4]:
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], inplace=True, log1p=True
)
[5]:
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
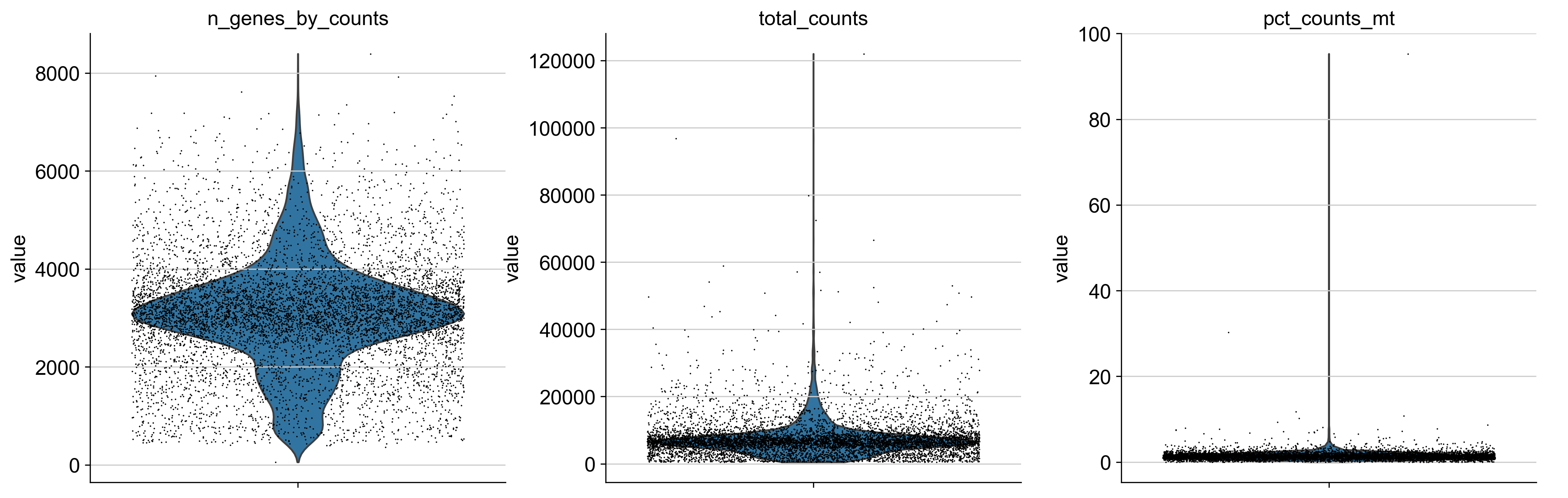
[6]:
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
[7]:
# Doublet detection using scrublet
sc.external.pp.scrublet(adata)
Automatically set threshold at doublet score = 0.24
Detected doublet rate = 3.0%
Estimated detectable doublet fraction = 39.0%
Overall doublet rate:
Expected = 5.0%
Estimated = 7.6%
[8]:
# Remove doublets + other QC metrics
adata = adata[adata.obs['predicted_doublet'] == False]
sc.pp.filter_cells(adata, max_genes = 6000)
sc.pp.filter_cells(adata, max_counts = 35000)
adata = adata[adata.obs['pct_counts_mt'] < 5]
Normalization, HVG, neighbors, PCA, UMAP#
[9]:
# Saving count data
adata.layers["counts"] = adata.X.copy()
# Normalization (Shifted logarithm)
sc.pp.normalize_total(adata, target_sum=None)
sc.pp.log1p(adata)
# scParadise use normalized data in adata.raw!!!
adata.raw = adata
# HVG
sc.pp.highly_variable_genes(adata, n_top_genes=2000)
# PCA
sc.tl.pca(adata)
# Nearest neighbors analysis
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=20)
# UMAP
sc.tl.umap(adata)
# Clusterization
sc.tl.leiden(adata, resolution=0.5, key_added='leiden_0.5')
[10]:
# Visualise predicted cell types levels and prediction probabilities
sc.pl.embedding(adata,
color = [
'leiden_0.5'
],
basis = 'X_umap',
frameon = False,
add_outline = True,
legend_loc = 'on data',
legend_fontsize = 9,
legend_fontoutline = 2,
save='clusterization.png')
WARNING: saving figure to file figures\X_umapclusterization.png
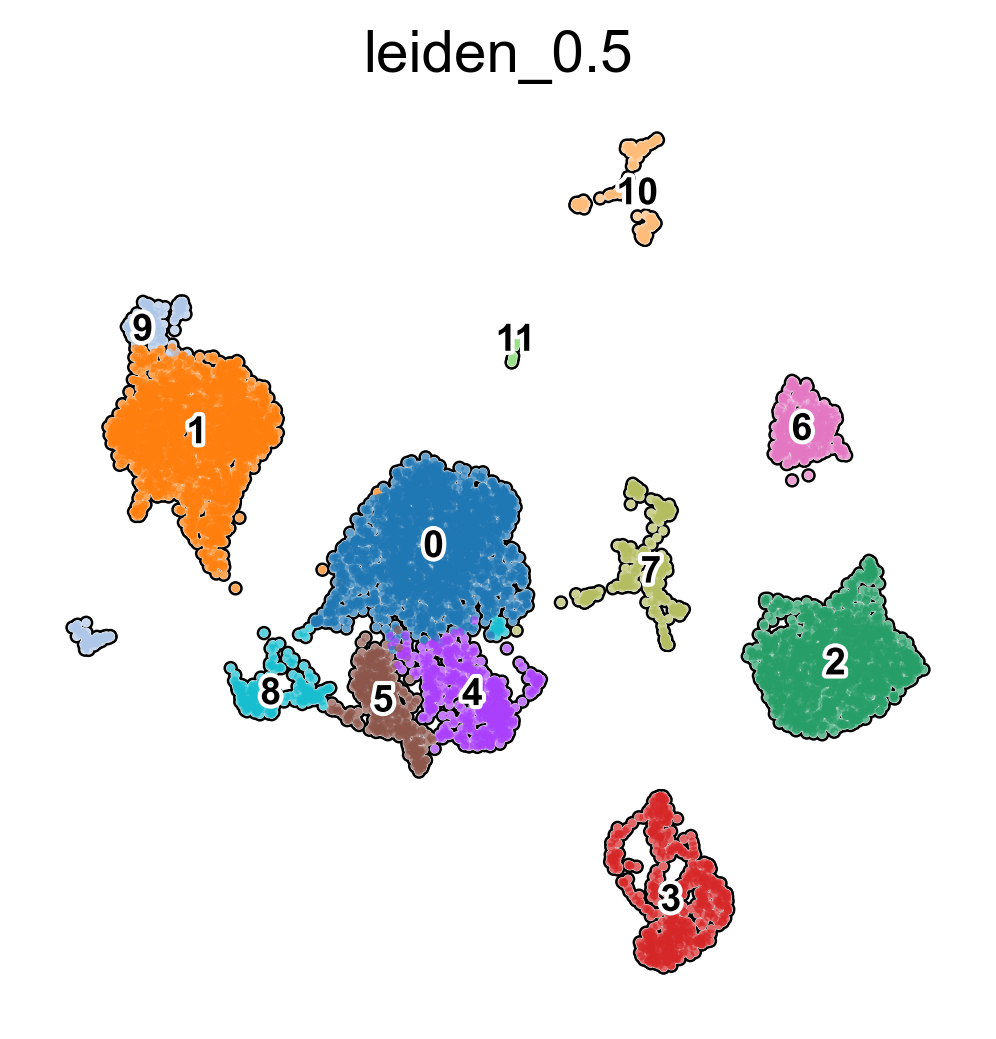
[11]:
adata.write_h5ad('mouse_clusterization.h5ad')
Observed ADT preprocessing#
[12]:
mu.prot.pp.clr(adata_adt)
[13]:
adata_adt = adata_adt[adata.obs_names]
adata_adt.obsm['X_umap'] = adata.obsm['X_umap']
[15]:
# Visualise naive T cells markers
sc.pl.embedding(adata_adt,
color = [
'Ms.CD3',
'Ms.CD4',
'Ms.CD8a',
'Ms.CD19',
'Ms.IgD',
'Ms.NK.1.1'
],
basis = 'X_umap',
frameon = False,
add_outline = True,
cmap = 'bwr',
legend_loc = 'on data',
legend_fontsize = 9,
legend_fontoutline = 2,
ncols = 3,
save='_observed_proteins.png'
)
WARNING: saving figure to file figures\X_umap_observed_proteins.png
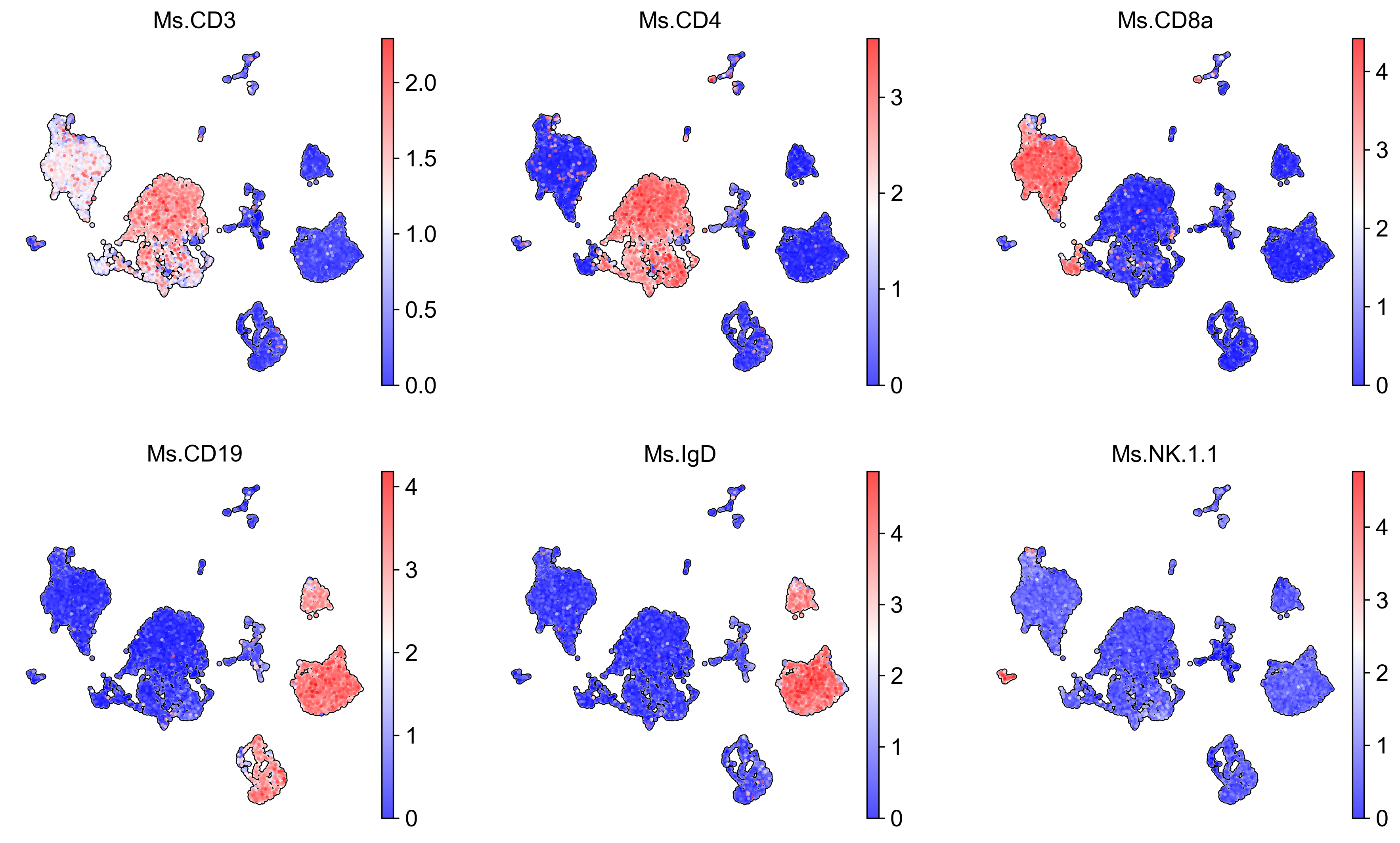
scParadise prediction (scEve)#
To use the model trained on human cells, it is necessary to replace the names of mouse genes with their orthologs from the human genome. Orthologs - genes in different species that evolved from a common ancestor by speciation (Example: Acta2 in mouse, ACTA2 in human).
[16]:
# Find orthologs of mouse genes in human genome
humanized_adata = translate(adata)
100%|██████████| 13167/13167 [00:18<00:00, 709.75it/s]
Found direct orthologs for 12219 genes.
Found multiple orthologs for 235 genes.
Found no orthologs for 442 genes.
Found no index in biomart for 271 genes.
100%|██████████| 235/235 [00:13<00:00, 17.83it/s]
[17]:
# Check matrix type and add it to raw
humanized_adata.X = sparse.csc_matrix(humanized_adata.X)
humanized_adata.raw = humanized_adata
[18]:
# Available models for cell type annotation
scparadise.sceve.available_models()
WARNING: RMSE, MASE, MSE are error metrics. Lower error metric value -> Better prediction.
RMSE - Root Mean Squared Error
MSE - Mean Squared Error
MAE - Mean Absolute Error
[18]:
| Tissue/Model name | Description | Suspension | RMSE | MAE | Number of Proteins | |
|---|---|---|---|---|---|---|
| 0 | Human_PBMC_3p | Peripheral blood mononuclear cells of healthy ... | cells | 0.305 | 0.226 | 224 |
| 1 | Human_PBMC_5p | Peripheral blood mononuclear cells of healthy ... | cells | 0.308 | 0.225 | 54 |
| 2 | Human_BMMC | Bone marrow mononuclear cell of healthy adults | cells | 0.706 | 0.454 | 134 |
[19]:
# Download model for modality prediction
scparadise.sceve.download_model(model_name = 'Human_PBMC_3p',
save_path = '')
[20]:
# Predict surface proteins using trained model
adata_adt_pred = scparadise.sceve.predict(humanized_adata,
path_model = 'Human_PBMC_3p_scEve/',
return_mdata = False)
Successfully loaded list of genes used for training model
Successfully loaded list of proteins used for training model
Successfully loaded model
[21]:
# Add UMAP from RNA data
adata_adt_pred.obsm['X_umap'] = adata.obsm['X_umap']
[22]:
# Visualise predicted proteins
sc.pl.embedding(adata_adt_pred,
color = [
'adt_CD3-1_pred',
'adt_CD4-1_pred',
'adt_CD8_pred',
'adt_CD19_pred',
'adt_IgD_pred',
'adt_CD56-2_pred'
],
basis = 'X_umap',
frameon = False,
add_outline = True,
cmap = 'bwr',
legend_loc = 'on data',
legend_fontsize = 9,
legend_fontoutline = 2,
ncols = 3,
save='_predicted_proteins.png'
)
WARNING: saving figure to file figures\X_umap_predicted_proteins.png
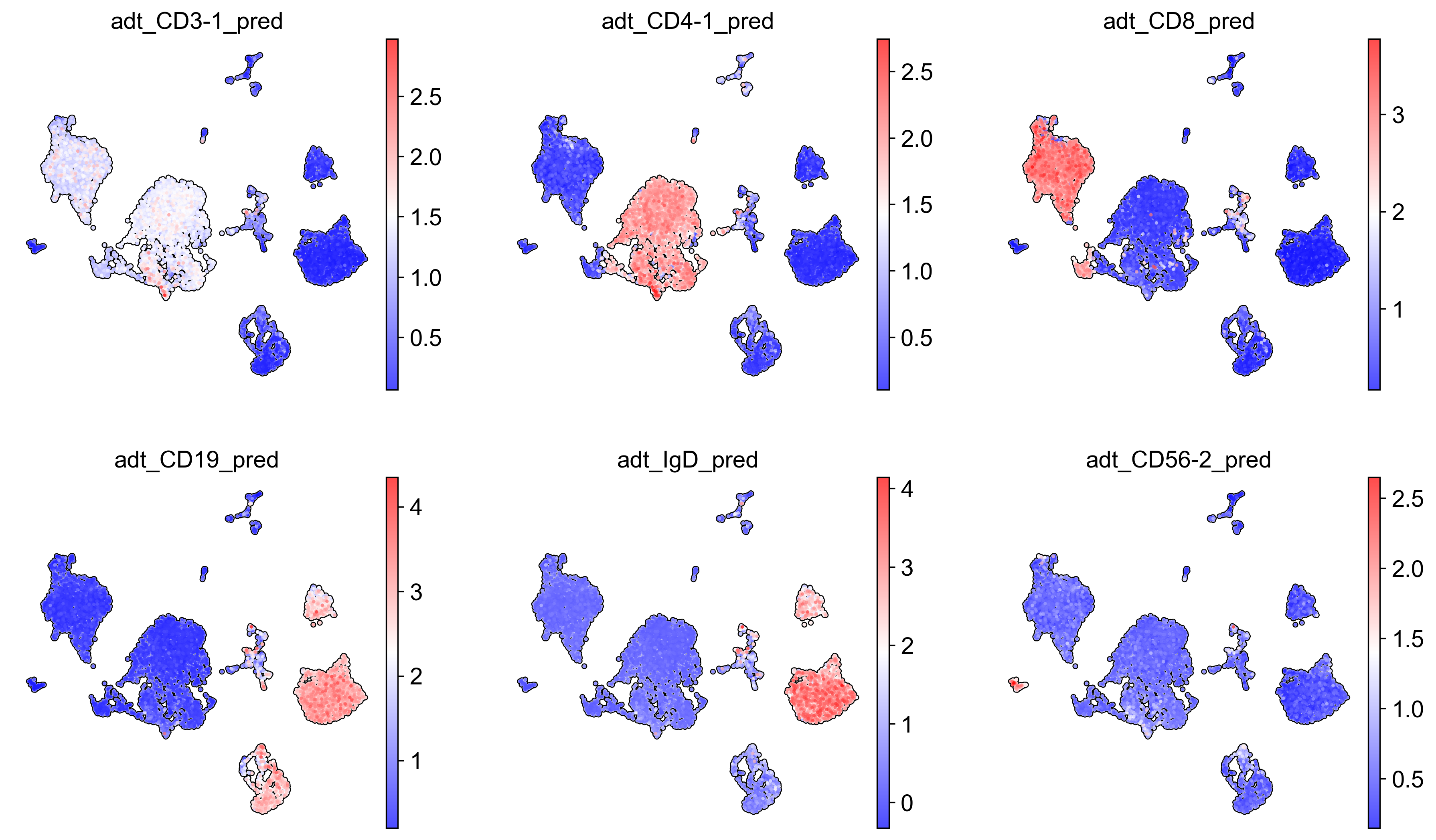
[ ]:
[23]:
pip list
Package Version
----------------------------- ---------------
absl-py 2.1.0
aiohttp 3.9.3
aiosignal 1.3.1
alabaster 0.7.16
alembic 1.13.2
anndata 0.10.5.post1
anndata2ri 1.3
annotated-types 0.6.0
annoy 1.17.3
ansicon 1.89.0
anyio 4.2.0
argon2-cffi 21.3.0
argon2-cffi-bindings 21.2.0
array_api_compat 1.4.1
arrow 1.3.0
astroid 2.14.2
asttokens 2.0.5
async-lru 2.0.4
async-timeout 4.0.3
atomicwrites 1.4.0
attrs 23.1.0
autopep8 2.0.4
Babel 2.11.0
backcall 0.2.0
backoff 2.2.1
bbknn 1.6.0
bcrypt 3.2.0
beautifulsoup4 4.12.2
binaryornot 0.4.4
black 24.8.0
bleach 4.1.0
blessed 1.20.0
boto3 1.34.37
botocore 1.34.37
celltypist 1.6.3
certifi 2024.8.30
cffi 1.16.0
chardet 4.0.0
charset-normalizer 2.0.4
chex 0.1.7
click 8.1.7
cloudpickle 3.0.0
colorama 0.4.6
colorlog 6.8.2
comm 0.1.2
commonmark 0.9.1
contextlib2 21.6.0
contourpy 1.2.0
cookiecutter 2.6.0
croniter 1.4.1
cryptography 43.0.0
cycler 0.12.1
Cython 3.0.8
dateutils 0.6.12
debugpy 1.6.7
decorator 5.1.1
deepdiff 6.7.1
defusedxml 0.7.1
Deprecated 1.2.14
diff-match-patch 20200713
dill 0.3.8
dm-tree 0.1.8
docrep 0.3.2
docstring-to-markdown 0.11
docutils 0.18.1
editor 1.6.6
et-xmlfile 1.1.0
etils 1.5.2
exceptiongroup 1.2.0
executing 0.8.3
fastapi 0.109.2
fastjsonschema 2.16.2
fbpca 1.0
filelock 3.13.1
flake8 7.0.0
flax 0.8.1
fonttools 4.48.1
frozenlist 1.4.1
fsspec 2024.2.0
geosketch 1.2
get-annotations 0.1.2
gprofiler-official 1.0.0
greenlet 3.0.3
h11 0.14.0
h5py 3.10.0
harmonypy 0.0.9
idna 3.4
igraph 0.10.8
imageio 2.36.0
imagesize 1.4.1
imbalanced-learn 0.12.3
importlib-metadata 7.0.1
importlib-resources 6.1.1
inflection 0.5.1
inquirer 3.2.3
intervaltree 3.1.0
ipykernel 6.28.0
ipython 8.15.0
isort 5.13.2
itsdangerous 2.1.2
jaraco.classes 3.2.1
jax 0.4.23
jaxlib 0.4.23
jedi 0.18.1
jellyfish 1.0.1
Jinja2 3.1.3
jinxed 1.2.1
jmespath 1.0.1
joblib 1.3.2
json5 0.9.6
jsonschema 4.19.2
jsonschema-specifications 2023.7.1
jupyter_client 8.6.0
jupyter_core 5.5.0
jupyter-events 0.8.0
jupyter-lsp 2.2.0
jupyter_server 2.10.0
jupyter_server_terminals 0.4.4
jupyterlab 4.0.11
jupyterlab-pygments 0.1.2
jupyterlab_server 2.25.1
jupytext 1.16.4
keyring 24.3.1
kiwisolver 1.4.5
lazy_loader 0.4
lazy-object-proxy 1.10.0
leidenalg 0.10.2
lightning 2.0.9.post0
lightning-cloud 0.5.64
lightning-utilities 0.10.1
llvmlite 0.42.0
louvain 0.8.1
Mako 1.3.5
markdown-it-py 2.2.0
MarkupSafe 2.1.3
matplotlib 3.8.2
matplotlib-inline 0.1.6
mccabe 0.7.0
mdit-py-plugins 0.4.2
mdurl 0.1.0
mistune 2.0.4
ml-collections 0.1.1
ml-dtypes 0.3.2
more-itertools 10.3.0
mousipy 0.1.6
mpmath 1.3.0
msgpack 1.0.7
mudata 0.2.3
multidict 6.0.5
multipledispatch 1.0.0
muon 0.1.6
mypy-extensions 1.0.0
natsort 8.4.0
nbclient 0.8.0
nbconvert 7.10.0
nbformat 5.9.2
nest-asyncio 1.5.6
networkx 3.2.1
notebook_shim 0.2.3
numba 0.59.0
numpy 1.24.4
numpydoc 1.7.0
numpyro 0.13.2
openpyxl 3.1.5
opt-einsum 3.3.0
optax 0.1.9
optuna 3.6.1
orbax-checkpoint 0.5.3
ordered-set 4.1.0
overrides 7.4.0
packaging 23.1
pandas 1.5.3
pandocfilters 1.5.0
paramiko 2.8.1
parso 0.8.3
pathspec 0.10.3
patsy 0.5.6
pexpect 4.8.0
pickleshare 0.7.5
pillow 10.2.0
pip 23.3.1
platformdirs 3.10.0
pluggy 1.0.0
ply 3.11
prometheus-client 0.14.1
prompt-toolkit 3.0.43
protobuf 4.25.2
psutil 5.9.0
ptyprocess 0.7.0
pure-eval 0.2.2
pycodestyle 2.11.1
pycparser 2.21
pydantic 2.1.1
pydantic_core 2.4.0
pydocstyle 6.3.0
pydot 2.0.0
pyflakes 3.2.0
Pygments 2.15.1
PyJWT 2.8.0
pylint 2.16.2
pylint-venv 3.0.3
pyls-spyder 0.4.0
PyNaCl 1.5.0
pynndescent 0.5.11
pyparsing 3.1.1
PyQt5 5.15.10
PyQt5-sip 12.13.0
PyQtWebEngine 5.15.6
pyro-api 0.1.2
pyro-ppl 1.8.6
python-dateutil 2.8.2
python-igraph 0.11.3
python-json-logger 2.0.7
python-lsp-black 2.0.0
python-lsp-jsonrpc 1.1.2
python-lsp-server 1.10.0
python-multipart 0.0.7
python-slugify 5.0.2
pytoolconfig 1.2.6
pytorch-lightning 2.2.0
pytorch-tabnet 4.1.0
pytz 2023.3.post1
pywin32 305.1
pywin32-ctypes 0.2.2
pywinpty 2.0.10
PyYAML 6.0.1
pyzmq 25.1.2
QDarkStyle 3.2.3
qstylizer 0.2.2
QtAwesome 1.3.1
qtconsole 5.5.1
QtPy 2.4.1
readchar 4.0.5
referencing 0.30.2
requests 2.31.0
rfc3339-validator 0.1.4
rfc3986-validator 0.1.1
rich 13.0.0
rope 1.12.0
rpds-py 0.10.6
rpy2 3.5.3
Rtree 1.0.1
runs 1.2.2
s3transfer 0.10.0
scanorama 1.7.4
scanpy 1.9.8
scib 1.1.4
scikit-image 0.24.0
scikit-learn 1.4.0
scikit-misc 0.3.1
scipy 1.12.0
scparadise 0.1.1b0
scrublet 0.2.3
scvi-tools 1.0.4
seaborn 0.13.2
Send2Trash 1.8.2
session-info 1.0.0
setuptools 68.2.2
sip 6.7.12
six 1.16.0
sniffio 1.3.0
snowballstemmer 2.2.0
sortedcontainers 2.4.0
soupsieve 2.5
sparse 0.15.1
SpeechRecognition 3.10.1
Sphinx 7.3.7
sphinxcontrib-applehelp 1.0.2
sphinxcontrib-devhelp 1.0.2
sphinxcontrib-htmlhelp 2.0.0
sphinxcontrib-jsmath 1.0.1
sphinxcontrib-qthelp 1.0.3
sphinxcontrib-serializinghtml 1.1.10
spyder 5.5.1
spyder-kernels 2.5.0
SQLAlchemy 2.0.31
stack-data 0.2.0
starlette 0.36.3
starsessions 1.3.0
statsmodels 0.14.1
stdlib-list 0.10.0
sympy 1.12
tabulate 0.9.0
tensorstore 0.1.53
terminado 0.17.1
text-unidecode 1.3
textdistance 4.2.1
texttable 1.7.0
threadpoolctl 3.2.0
three-merge 0.1.1
tifffile 2024.8.30
tinycss2 1.2.1
tomli 2.0.1
tomlkit 0.13.2
toolz 0.12.1
torch 2.2.0
torchmetrics 1.3.0.post0
tornado 6.3.3
tqdm 4.66.1
traitlets 5.7.1
types-python-dateutil 2.8.19.20240106
typing_extensions 4.9.0
tzdata 2023.4
tzlocal 5.2
ujson 5.10.0
umap-learn 0.5.5
Unidecode 1.3.8
urllib3 1.26.18
uvicorn 0.27.0.post1
watchdog 4.0.1
wcwidth 0.2.5
webencodings 0.5.1
websocket-client 0.58.0
websockets 12.0
whatthepatch 1.0.2
wheel 0.41.2
wrapt 1.16.0
xarray 2024.1.1
xmod 1.8.1
yapf 0.40.2
yarl 1.9.4
zipp 3.17.0
Note: you may need to restart the kernel to use updated packages.
[ ]: