Unknown cell type identification using scAdam#
scAdam is specifically developed for annotating cell types, especially focusing on rare cell types that may be underrepresented in the dataset.
Advantages
scAdam not only detects all cell types in any test dataset but also generates reproducible results, which is an important aspect for reliable biological interpretation.
It enables multitasking by allowing the researchers to extract individual cell types for targeted investigations.
Unknown cell type identification makes it possible to identify new cell types that are absent from the data on which the scAdam model was trained. In addition, this enables cross-tissue cell type annotation, as demonstrated in this tutorial.
Integration with Other Tools: scAdam is part of a bigger toolkit that includes other tools, such as scEve for surface protein prediction and scNoah for benchmarking, making it a comprehensive solution for single-cell analysis.
[1]:
# Python packages
import warnings
warnings.simplefilter('ignore')
import scanpy as sc
import scparadise
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
sc.set_figure_params(dpi = 80)
[2]:
# Download example dataset from Figshare:
# https://figshare.com/ndownloader/files/62994628
# Use wget or simply paste the link into your browser to download.
[3]:
# Load visceral adipose tissue dataset
adata = sc.read_h5ad('VAT_sample.h5ad')
adata
[3]:
AnnData object with n_obs × n_vars = 16520 × 28137
obs: 'sample', 'ground_truth'
obsm: 'X_harmony', 'X_pca', 'X_umap'
[4]:
# Saving count data
adata.layers['counts'] = adata.X.copy()
# Normalizing to median total counts
sc.pp.normalize_total(adata)
# Logarithmize the data
sc.pp.log1p(adata)
[5]:
# Available models for cell type annotation
df = scparadise.scadam.available_models()
# Show models related to humans
df_human = df[df['Tissue/Model name'].str.startswith('Human_')]
df_human
[5]:
| Tissue/Model name | Description | Suspension | Accuracy | Balanced Accuracy | Number of Levels | |
|---|---|---|---|---|---|---|
| 0 | Human_BMMC | Bone marrow mononuclear cell of healthy adults | cells | 0.947 | 0.942 | 3 |
| 1 | Human_Bone_Marrow | A Balanced Bone Marrow Reference | cells | 0.881 | 0.861 | 3 |
| 2 | Human_Brain_atlas | Human Brain Cell Atlas v1.0 | nuclei | 0.998 | 0.998 | 2 |
| 3 | Human_Brain_SEA_AD | Seattle Alzheimer’s Disease Brain Cell Atlas | nuclei | 0.997 | 0.997 | 3 |
| 4 | Human_CC_Dev_RNA | Multi-omic profiling of the developing human c... | nuclei | 0.974 | 0.975 | 2 |
| 5 | Human_CC_Dev_ATAC | Multi-omic profiling of the developing human c... | nuclei | 0.916 | 0.912 | 2 |
| 6 | Human_Heart | Human heart CITE-seq analysis of healthy and d... | cells | 0.957 | 0.956 | 2 |
| 7 | Human_Kidney_cell | scRNA-seq of the Adult Human Kidney (V. 1.5) | cells | 0.974 | 0.974 | 3 |
| 8 | Human_Kidney_nucleus | snRNA-seq of the Adult Human Kidney (V. 1.5) | nuclei | 0.973 | 0.972 | 3 |
| 9 | Human_Lung | Core Human Lung Cell Atlas | cells | 0.965 | 0.964 | 5 |
| 10 | Human_Lung_Cancer | Extended single-cell lung cancer atlas (LuCA) | cells | 0.937 | 0.936 | 3 |
| 11 | Human_oropharyngeal_SCC | Oropharyngeal HPV+/HPV- squamous cell carcinom... | cells | 0.972 | 0.968 | 2 |
| 12 | Human_Pancreas | Pancreatic islet atlas | cells | 0.996 | 0.989 | 1 |
| 13 | Human_PBMC | Peripheral blood mononuclear cells of healthy ... | cells | 0.979 | 0.979 | 3 |
| 14 | Human_Retina_cell | Single cell atlas of the human retina | cells | 0.984 | 0.979 | 4 |
| 15 | Human_Retina_nucleus | Single nucleus atlas of the human retina | nuclei | 0.994 | 0.994 | 2 |
| 16 | Human_Subcutaneous_AT | Subcutaneous adipose tissue atlas | cells | 0.973 | 0.954 | 3 |
| 17 | Human_Testes | Single cell atlas of the human testes | cells | 0.991 | 0.991 | 2 |
| 18 | Human_Visceral_AT | Visceral adipose tissue atlas | cells | 0.978 | 0.975 | 3 |
[6]:
# Download model for cell type prediction
scparadise.scadam.download_model('Human_Subcutaneous_AT', save_path='')
[7]:
# Predict cell types using scAdam model with unknown cell type identification
adata = scparadise.scadam.predict(
adata,
path_model = 'Human_Subcutaneous_AT_scAdam',
detect_unknown = True # Turn on unknown cell type identification
)
scAdam model with unknown detector loaded from Human_Subcutaneous_AT_scAdam
Gene alignment:
Model features: 1198
Matched features: 1197 (99.9%)
Predicting: 100%|██████████████████████████████████████████████████████████████████████| 65/65 [00:00<00:00, 114.22it/s]
Gradient threshold calculated using Otsu method: 1.5836894512176514
Entropy threshold calculated using Otsu method: 2.084172487258911
Distance threshold calculated using Otsu method: 9.879642486572266
Unknown cell type detection using 3 methods. Threshold: 2 votes.
Detected 3479 unknown cells (21.1%)
Added cell type column: pred_celltype_l1
Added probabilities column: pred_celltype_l1_probability
Added cell type column: pred_celltype_l2
Added probabilities column: pred_celltype_l2_probability
Added cell type column: pred_celltype_l3
Added probabilities column: pred_celltype_l3_probability
[8]:
# Sort order cells
adata = adata[adata.obs.sort_values("pred_unknown").index]
All scores work on the same principle: The higher the value, the more confident the model is that it doesn’t know this cell’s type.
[9]:
# Visualise prediction results with unknown cell type scores (gradient, entropy, distance)
sc.pl.umap(
adata,
color = [
'pred_celltype_l1',
'pred_celltype_l2',
'pred_unknown',
'gradient_score', # unknown cell type score
'entropy_score', # unknown cell type score
'distance_score' # unknown cell type score
],
frameon = False,
legend_fontsize = 8,
ncols = 3,
cmap = 'bwr',
vmin = 'p01',
vmax = 'p99'
)
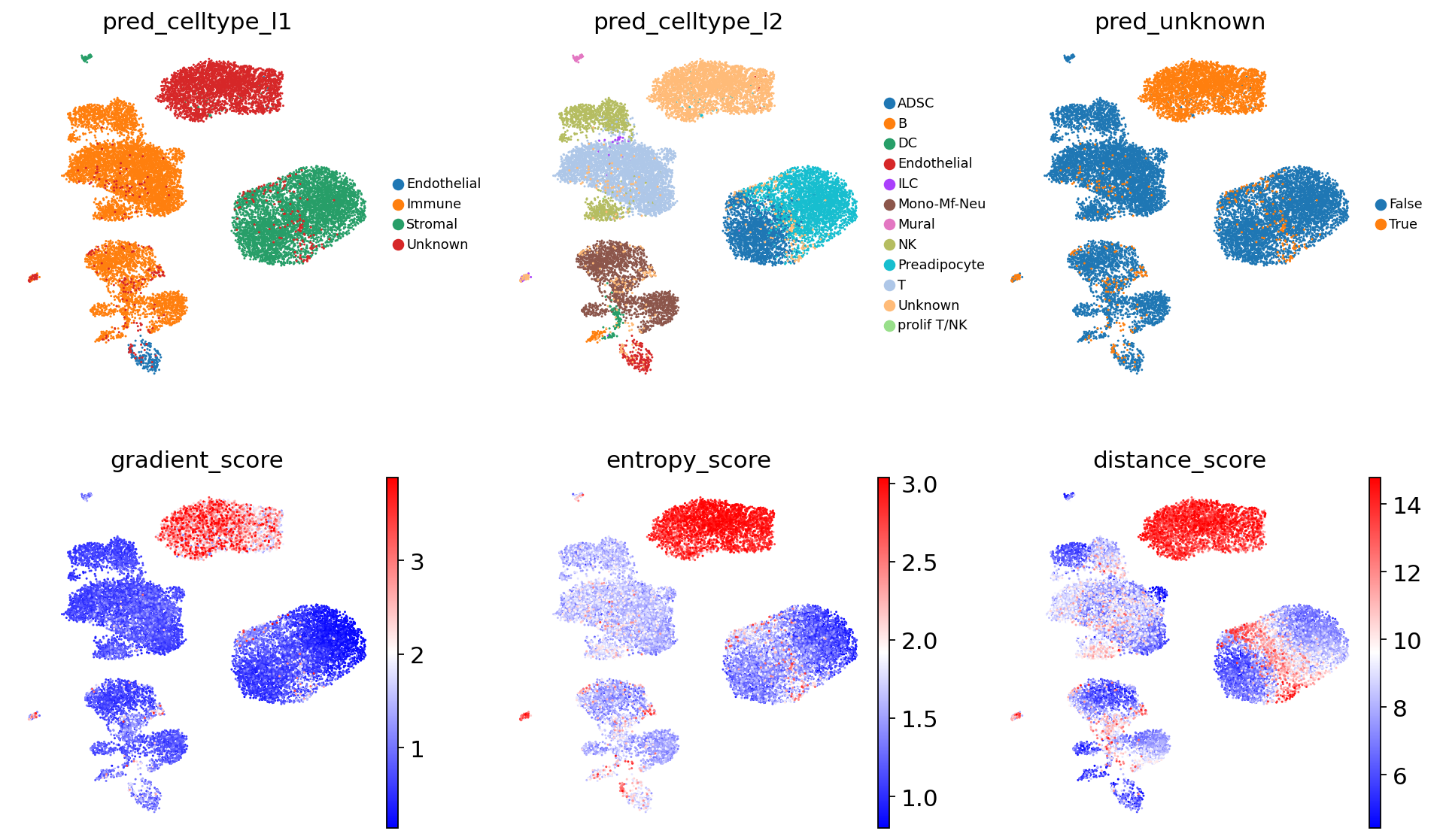
Analysis of the scores indicates the presence of two additional cell types in visceral adipose tissue that are absent in subcutaneous adipose tissue.
The dataset already contains a ground-truth cell type annotation, produced independently of scAdam predictions (using marker genes). To compare the ground-truth cell annotations with the predicted ones, we will recolor the cells using the same colors for ground-truth and predicted cell type annotations.
[10]:
# Define colors
color_map = {
"ADSC": "#1f77b4",
"T": "#ff7f0e",
"NK": "#279e68",
"ILC": "#d62728",
"Mural": "#aa40fc",
"Mono-Mf-Neu": "#8c564b",
"Mesothelial": "#b5bd61",
"Preadipocyte": "#17becf",
"B": "#aec7e8",
"Endothelial": "#ffbb78",
"Mast": "#98df8a",
"prolif T/NK": "#ff9896",
"Unknown": "#c5b0d5"
}
default_color = "#bdbdbd"
# Recolor cells
celltype_col = 'ground_truth'
cats = list(adata.obs[celltype_col].cat.categories)
adata.uns[f"{celltype_col}_colors"] = [color_map.get(c, default_color) for c in cats]
celltype_col = 'pred_celltype_l2'
cats = list(adata.obs[celltype_col].cat.categories)
adata.uns[f"{celltype_col}_colors"] = [color_map.get(c, default_color) for c in cats]
[11]:
# Visualize ground_truth vs pred_celltype_l2
sc.pl.umap(
adata,
color = [
'ground_truth',
'pred_celltype_l2',
'pred_unknown',
'gradient_score',
'entropy_score',
'distance_score'
],
frameon = False,
legend_fontsize = 8,
ncols = 3,
cmap = 'bwr',
vmin = 'p01',
vmax = 'p99'
)
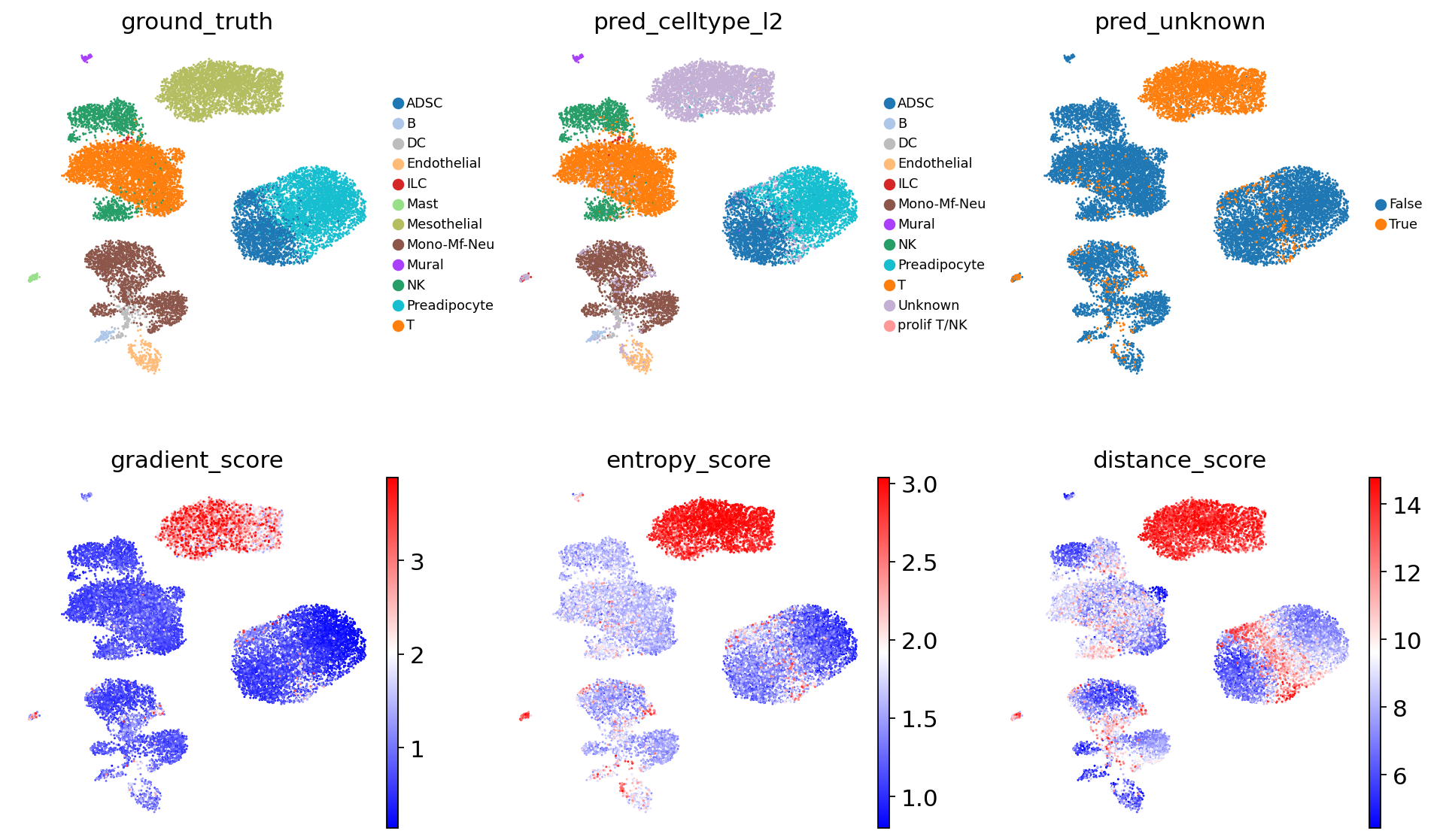
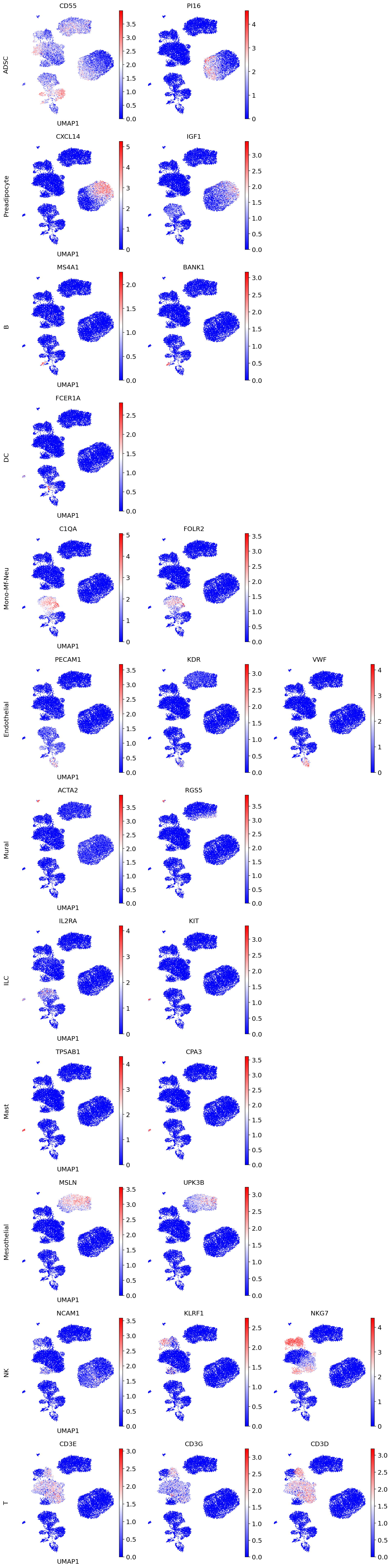
We recommend validating scAdam cell type prediction using the corresponding marker genes.
[12]:
marker_genes = {
"ADSC": ['CD55', 'PI16'],
"Preadipocyte": ['CXCL14', 'IGF1'],
"B": ['MS4A1', 'BANK1'],
"DC": ['FCER1A'],
"Mono-Mf-Neu": ['C1QA', 'FOLR2'],
"Endothelial": ['PECAM1', 'KDR', 'VWF'],
"Mural": ['ACTA2', 'RGS5'],
"ILC": ['IL2RA', 'KIT'],
"Mast": ['TPSAB1', 'CPA3'],
"Mesothelial": ['MSLN', 'UPK3B'],
"NK": ['NCAM1', 'KLRF1', 'NKG7'],
"T": ['CD3E', 'CD3G', 'CD3D'],
}
[13]:
# Make Axes
# Number of needed rows and columns (based on the row with the most columns)
nrow = len(marker_genes)
ncol = max([len(vs) for vs in marker_genes.values()])
fig, axs = plt.subplots(nrow, ncol, figsize=(4 * ncol, 4 * nrow))
# Plot expression for every marker on the corresponding Axes object
for row_idx, (cell_type, markers) in enumerate(marker_genes.items()):
col_idx = 0
for marker in markers:
ax = axs[row_idx, col_idx]
sc.pl.umap(
adata,
color=marker,
ax=ax,
show=False,
cmap = 'bwr',
frameon=False
)
# Add cell type as row label - here we simply add it as ylabel of
# the first Axes object in the row
if col_idx == 0:
# We disabled axis drawing in UMAP to have plots without background and border
# so we need to re-enable axis to plot the ylabel
ax.axis("on")
ax.tick_params(
top="off",
bottom="off",
left="off",
right="off",
labelleft="on",
labelbottom="off",
)
ax.set_ylabel(cell_type + "\n", rotation=90, fontsize=14)
ax.set(frame_on=False)
col_idx += 1
# Remove unused column Axes in the current row
while col_idx < ncol:
axs[row_idx, col_idx].remove()
col_idx += 1
# Alignment within the Figure
fig.tight_layout()
[14]:
adata.write_h5ad('adata_predicted_with_unknown.h5ad')
[17]:
import session_info
session_info.show()
[17]:
Click to view session information
----- anndata 0.11.4 matplotlib 3.10.8 numpy 2.2.6 pandas 2.3.3 scanpy 1.11.5 scparadise 1.0.0 session_info v1.0.1 -----
Click to view modules imported as dependencies
81d243bd2c585b0f4821__mypyc NA PIL 12.1.1 aiohappyeyeballs 2.6.1 aiohttp 3.13.3 aiosignal 1.4.0 anyio NA arrow 1.4.0 asttokens NA async_timeout 5.0.1 attr 26.1.0 attrs 26.1.0 babel 2.18.0 certifi 2026.02.25 cffi 2.0.0 charset_normalizer 3.4.6 cloudpickle 3.1.2 colorlog NA comm 0.2.3 cuda 12.9.4 cycler 0.12.1 cython_runtime NA dateutil 2.9.0.post0 debugpy 1.8.20 decorator 5.2.1 defusedxml 0.7.1 exceptiongroup 1.3.1 executing 2.2.1 fastjsonschema NA fqdn NA frozenlist 1.8.0 fsspec 2026.2.0 h5py 3.16.0 idna 3.11 imblearn 0.14.1 ipykernel 7.2.0 isoduration NA jedi 0.19.2 jinja2 3.1.6 joblib 1.5.3 json5 0.13.0 jsonpointer 3.1.0 jsonschema 4.26.0 jsonschema_specifications NA jupyter_events 0.12.0 jupyter_server 2.17.0 jupyterlab_server 2.28.0 kiwisolver 1.5.0 lark 1.3.1 lazy_loader 0.5 legacy_api_wrap NA llvmlite 0.46.0 markupsafe 3.0.3 matplotlib_inline 0.2.1 mpl_toolkits NA mudata 0.3.3 multidict 6.7.1 muon 0.1.7 natsort 8.4.0 nbformat 5.10.4 numba 0.64.0 optuna 4.8.0 overrides NA packaging 25.0 parso 0.8.6 patsy 1.0.2 pexpect 4.9.0 platformdirs 4.9.4 plottable 0.1.5 prometheus_client NA prompt_toolkit 3.0.52 propcache 0.4.1 psutil 7.2.2 ptyprocess 0.7.0 pure_eval 0.2.3 pycparser 3.00 pydev_ipython NA pydevconsole NA pydevd 3.2.3 pydevd_file_utils NA pydevd_plugins NA pydevd_tracing NA pygments 2.19.2 pynndescent 0.6.0 pyparsing 3.3.2 pythonjsonlogger NA pytorch_tabnet NA pytz 2026.1.post1 referencing NA requests 2.32.5 rfc3339_validator 0.1.4 rfc3986_validator 0.1.1 rfc3987_syntax NA rpds NA scipy 1.15.3 seaborn 0.13.2 send2trash NA shap 0.49.1 six 1.17.0 skimage 0.25.2 sklearn 1.7.2 sklearn_compat 0.1.5 slicer NA stack_data 0.6.3 statsmodels 0.14.6 threadpoolctl 3.6.0 torch 2.10.0+cu128 torchgen NA tornado 6.5.5 tqdm 4.67.3 traitlets 5.14.3 typing_extensions NA umap 0.5.11 uri_template NA urllib3 2.6.3 wcwidth 0.6.0 webcolors NA websocket 1.9.0 yaml 6.0.3 yarl 1.23.0 zmq 27.1.0 zoneinfo NA
----- IPython 8.38.0 jupyter_client 8.8.0 jupyter_core 5.9.1 jupyterlab 4.5.6 ----- Python 3.10.20 (main, Mar 11 2026, 17:46:40) [GCC 14.3.0] Linux-6.6.87.2-microsoft-standard-WSL2-x86_64-with-glibc2.39 ----- Session information updated at 2026-03-22 14:36